Your new post is loading...
Your new post is loading...

|
Scooped by
Gilbert C FAURE
December 27, 2013 5:57 AM
|
Pathology, Diagnosis and Therapy
|
|
Scooped by
Gilbert C FAURE
June 28, 4:46 AM
|
Wonderful news!
Five lupus patients in England are in remission after being treated with a revolutionary therapy that genetically modifies their own cells, in a medical breakthrough that could offer people a cure, doctors have said.
CAR (chimeric antigen receptor) T-cell therapy involves removing a type of white blood cell also called T lymphocytes, which are crucial for hunting out infected or damaged cells, and engineering them to spot and destroy disease. The T-cells are then fed back into the patient via an infusion to reset their immune system.
The therapy is already revolutionising cancer treatment. Now medics in London have successfully used the technique to effectively cure five NHS patients with severe lupus aged between 19 and 50.
CAR T-cell therapy, which patients only need to have once, could transform lupus treatment and remove the need for lifelong medication, doctors said.
|
|
Scooped by
Gilbert C FAURE
June 19, 1:19 PM
|
Sjögren’s Syndrome (SjS) has historically been associated with classical anti-Ro60/SSA, Ro52/SSA and La/SSB, however they are lacking in one third of the patients, which induces delays in diagnosis, and their disease-contributing role is debated. Here we have applied a SjS-tailored Systems Serology approach to a cohort of 58 SjS and 16 non-SjS sicca syndrome patients, and 40 healthy individuals, involving a multiplex assay measuring antibody isotype, subclass, Fc Receptor and complement engagement to 14 SjS-related autoantigens, an antibody-glycosylation profiling assay and a phagocytosis cell-based assay. Via a machine learning approach, we have identified unique autoantibody signatures, including classical and non-classical autoantigens-related features especially involving autoantigen-specific Fc Receptor binding, with apparent functional consequences. These findings provide interesting insights into the autoantibody responses in SjS, possibly paving the way for improved diagnostics, especially in difficult-to-diagnose patients (e.g., seronegative SjS and non-SjS sicca syndrome patients), and novel therapeutic options targeting autoantibody-specific Fc/Fc Receptor-related effector functions.
|
|
Scooped by
Gilbert C FAURE
June 8, 4:10 AM
|
🎾 Dimanche, Alexander Zverev a soulevé la Coupe des Mousquetaires 🏆.
Ce que les caméras n’ont pas filmé, c’est le deuxième match qu’il jouait en même temps. Celui contre sa propre glycémie.
Zverev est diabétique de type 1 depuis l’âge de 3 ans et demi. Une maladie auto-immune qui détruit les cellules du pancréas produisant l’insuline.
Résultat : son corps n’en fabrique plus une seule goutte. Sans injection, ce n’est pas une gêne,c’est un pronostic vital engagé.
Alors imaginez gérer cela pendant une finale de plus de 4 heures.
L’effort prolongé fait chuter la glycémie : le muscle consomme le glucose, la sensibilité à l’insuline grimpe. Mais le stress, l’adrénaline et le cortisol de la compétition la font, eux, remonter. Ajoutez la chaleur de juin sur l’ocre parisienne, qui accélère l’absorption de l’insuline et peut fausser les capteurs. La glycémie part dans les deux sens, sans prévenir.
Comment fait-il ?
Pas de pompe dernier cri. Zverev gère avec un capteur de glycémie en continu,qu’il consulte à chaque changement de côté et un simple stylo à insuline qu’il dégaine sur le court. Il a d’ailleurs dû batailler pour en obtenir le droit à Roland-Garros, qui le lui refusait au départ. « Si je ne le fais pas, ma vie est en danger », a-t-il répondu.
On lui avait pourtant dit, enfant, qu’une carrière de haut niveau serait impossible. Des médecins, « derrière leur bureau ». Ils avaient tort.
Et c’est là que la science a rattrapé le dogme. À condition de maintenir la glycémie dans la cible, une personne diabétique de type 1 peut atteindre une capacité aérobie comparable à celle d’une personne non diabétique. Le consensus international de référence (Riddell, The Lancet Diabetes & Endocrinology, 2017) en donne la recette :
- réduire l’insuline avant l’effort sans jamais la couper
- apporter 30 à 60 g de glucides par heure
- lire non pas le chiffre, mais la tendance 📈.
Team Novo Nordisk, équipe cycliste professionnelle composée exclusivement de diabétiques de type 1, le prouve chaque saison dans le peloton international.
Soyons clairs, en cardiologie/diabétologie : on ne « gagne » pas contre un diabète de type 1. La maladie est là le lendemain matin. Elle reste un facteur de risque cardiovasculaire majeur, à surveiller toute une vie.
Ce que Zverev démontre, ce n’est pas que la volonté guérit. C’est que la technologie et la discipline déplacent le plafond,celui que des décennies de prudence médicale avaient fixé bien trop bas.
Aujourd’hui, on gère le diabète de type 1. Demain, en guérira-t-on ?
Les avancées en cours méritent un post à part,j’y reviens bientôt.
À 9 ans, on lui a listé tout ce qu’il ne ferait jamais.
Dimanche, pendant que le public suivait le score, lui suivait une autre courbe,sur son capteur, entre deux jeux.
Les deux ont fini dans le vert.
Hôpital Américain de Paris
|
|
Scooped by
Gilbert C FAURE
May 13, 3:29 AM
|
Antiphospholipid antibodies (aPL) are directed against phospholipids and phospholipid-binding proteins. Laboratory assays used to detect aPL include serological tests for aPL against β2-glycoprotein 1, cardiolipin and other molecules, as well as functional assays for lupus anticoagulant. The presence of aPL can lead to endothelial dysfunction or a hypercoagulable state through prothrombotic and antifibrinolytic mechanisms. These processes, often in conjunction with a ‘second hit’, such as trauma, surgery, or other causes of hypercoagulability or stasis, can lead to venous or arterial thrombosis. The thrombotic risk associated with aPL is best recognized in thrombotic antiphospholipid syndrome, characterized by a persistently positive test for lupus anticoagulant or seropositivity for aPL associated with venous, arterial or microvascular thrombosis. However, aPL seropositivity and its clinical effect on thrombotic events have been increasingly recognized in a broader group of individuals who do not meet traditional research criteria for thrombotic antiphospholipid syndrome. In this Review, we provide an overview of the evidence related to aPL seropositivity in individuals with or without previous thrombosis and the clinical relevance of aPL seropositivity in predicting the risk of thrombotic cardiovascular events. We discuss potential management strategies and identify key knowledge gaps that warrant further research. Antiphospholipid antibodies (aPL) are associated with an elevated risk of thromboembolic events in patients with antiphospholipid syndrome and, increasingly, in those without previous thrombosis. In this Review, Bikdeli and colleagues discuss the clinical relevance of aPL seropositivity in predicting the risk of thrombotic cardiovascular events, summarize potential management strategies and identify key knowledge gaps that warrant further research.
|
|
Scooped by
Gilbert C FAURE
May 9, 5:28 AM
|
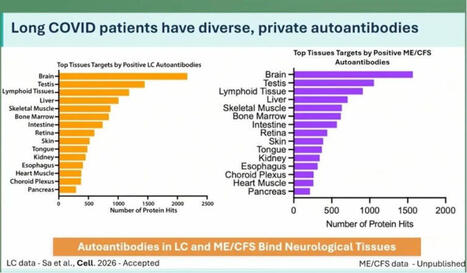
Help!! They're attacking our balls!!
At the ME/CFS conference in Berlin today, Keyla Sá from the Akiko Iwasaki group at Yale showed that Long Covid patients have more autoantibodies targeting neurological tissue. This was apparently tested in both human and mouse tissue.
In ME there are even more autoantibodies than in Long Covid, but number 2 on the list are the cojones.
Interesting. 🍒
Maybe this helps explain why some men develop ME even though the disease is normally more common in women?
Testosterone seems to actually protect against this kind of immune misfire. We see this in research on trans men: https://lnkd.in/ewjxZsRZ
If that protection drops for any reason, the immune system can start attacking your own tissues?
Including your balls?
That’s bullocks!!
https://lnkd.in/eE-giXTv
#pwme #myalgicE #millionsmissing #severeME #MECFSConf2026
|
|
Scooped by
Gilbert C FAURE
April 10, 3:59 AM
|
L'importance du thymus de plus en plus reconnue.
On est loin d'un organe qui n'avait plus d'importance après la puberté.
|
|
Scooped by
Gilbert C FAURE
March 26, 4:13 AM
|
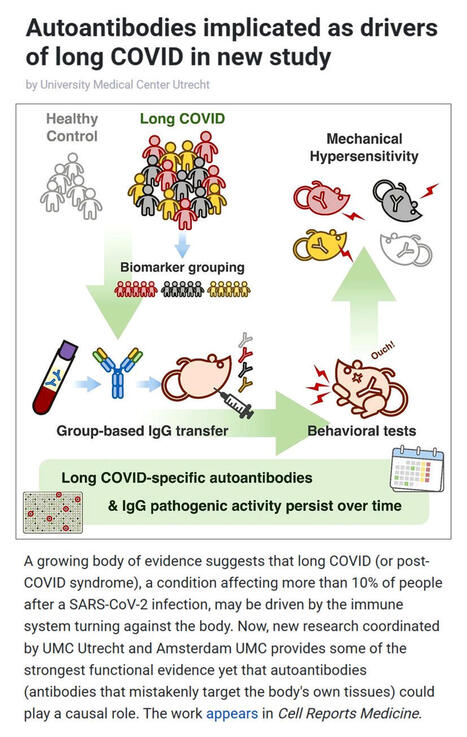
A new study coordinated by UMC Utrecht and Amsterdam UMC provides compelling evidence that #longCOVID may be driven by #autoimmunity - where the #immunesystem turns against the body.
▪️ Affecting over 10% of people after #SARSCoV2 #infection, long COVID is known for symptoms like fatigue, pain, and cognitive dysfunction. Yet its underlying #biology has remained unclear.
▪️In this study, researchers transferred IgG #antibodies from long COVID patients into mice triggering persistent pain-like symptoms that lasted weeks. Remarkably, antibodies collected from the same patients two years later produced the same effect, suggesting a long-lasting disease mechanism.
▪️Co-study leads Prof. Niels Eijkelkamp (UMC Utrecht) and Dr. Jeroen den Dunnen (Amsterdam UMC) highlight two key insights:
- Long COVID may be driven by persistent, pathogenic autoantibodies
- The condition is likely heterogeneous, with distinct biological subtypes
▪️The team also identified diverse autoantibodies targeting immune, neurological, and metabolic pathways - many persisting for years.
▪️These findings, published in Cell Reports Medicine by Cell Press, not only strengthen the case for an autoimmune basis of long COVID but also open the door to targeted treatments like immunoadsorption, plasmapheresis, and precision immunotherapy.
As research converges globally on similar findings, this marks a major step toward understanding - and treating - long COVID.
🗃️ See comments for reference.
|
|
Scooped by
Gilbert C FAURE
January 21, 4:18 AM
|
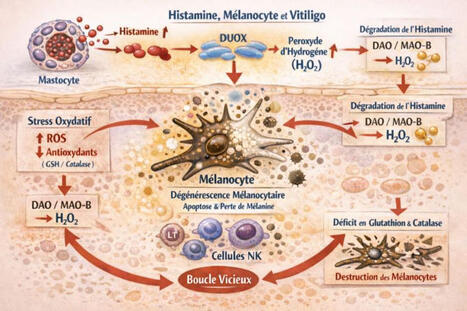
🔬 Histamine, mélanocytes et vitiligo : et si le chaînon manquant était oxydatif ?
Le vitiligo est encore trop souvent présenté comme une maladie exclusivement auto-immune.
Pourtant, les données accumulées depuis plusieurs années dessinent une lecture plus intégrée, où stress oxydatif, neuro-inflammation et biologie de l’histamine jouent un rôle central.
🧬 Le mélanocyte : une cellule particulièrement vulnérable
Le mélanocyte est une cellule à haute activité métabolique, exposée en permanence à des espèces réactives de l’oxygène (ROS), notamment via la mélanogenèse. Sa survie dépend étroitement de l’équilibre redox local.
🧠 L’histamine : bien plus qu’un médiateur allergique
Dans la peau, l’histamine est libérée par les mastocytes, mais aussi modulée par les kératinocytes, les fibres nerveuses et l’immunité innée.
Elle agit comme un amplificateur inflammatoire et oxydatif, en particulier via l’activation des enzymes DUOX, productrices de peroxyde d’hydrogène (H₂O₂) et sa propre dégradation (DAO en extracellulaire, MAO-B en intracellulaire), qui génère elle aussi du H₂O₂.
Un paradoxe peu discuté
Même lorsque l’histamine est “correctement” dégradée, elle augmente la charge oxydative locale. Si les systèmes antioxydants, notamment glutathion et catalase sont insuffisants, le résultat est une accumulation de H₂O₂ toxique pour le mélanocyte.
🔁 Une boucle auto-entretenue
Histamine ↑ → ROS ↑ → stress oxydatif → souffrance mélanocytaire → signaux de danger → activation immunitaire → mastocytes → histamine ↑
🎯 Implication clinique majeure
Le vitiligo peut être relu comme une pathologie de déséquilibre histamino-oxydatif local, où l’auto-immunité apparaît souvent comme une conséquence plus que comme le point de départ.
👉 Cette approche ouvre des pistes complémentaires comme la réduction de la charge histaminique, la protection antioxydante ciblée, la modulation mastocytaire et la prise en compte du terrain neuro-immun et métabolique.
🔍 Changer de prisme, c’est parfois changer le pronostic.
Dr Lucie WETCHOKO
Source de réflexion:
1- constats de consultation
2- Rôle de l'histamine comme médiateur toxique dans la pathogenèse du vitiligo: 10.4103/0019-5154.119947
|
|
Scooped by
Gilbert C FAURE
January 14, 4:11 AM
|
Vitiligo is an autoimmune disease of melanocyte destruction, which manifests as progressive, patchy loss of pigmentation in the skin. As one of most common autoimmune diseases, vitiligo inflicts a significant psychosocial burden. Research over the past two decades has revealed the underlying immune mechanisms of vitiligo, with key studies combining detailed analyses of patient tissue samples with mechanistic experiments in mouse models. Vitiligo has emerged as a prototypical CD8+ T cell-mediated autoimmune disease, with cooperation between innate immune cells, dendritic cells, T cells, keratinocytes and fibroblasts driving autoimmune pathology against the uniquely susceptible melanocyte target. The study of vitiligo has also revealed aspects of CD8+ T cell memory and resident memory against self-antigens. This work has drawn from, and contributed to, the study of melanoma immunology. Whereas drugs used for other autoimmune conditions have been largely ineffective in treating vitiligo, a growing base of knowledge recently led to the first successful FDA-approved immune-modulating drugs for vitiligo. This review focuses on the immunology of vitiligo: the mechanisms that drive melanocyte destruction, the biology of aberrant T cell responses against melanocytes and therapeutic means for counteracting this autoimmune condition. This Review from Turk and Huang discusses the immune processes involved in the development of vitiligo, an autoimmune disease in which melanocyte destruction causes loss of skin pigmentation. The authors highlight key studies from the past two decades that have shaped our understanding of vitiligo and led to newly approved immune-modulating drugs for the disease.
|
|
Scooped by
Gilbert C FAURE
December 29, 2025 9:16 AM
|
Autoimmunity, decoded at single-cell resolution: toward mechanism-driven therapies and smarter diagnostics
Autoimmune diseases are often treated as distinct clinical entities, yet patients experience overlapping biology, variable responses to therapy, and chronic immune dysregulation. This new large-scale single-cell study delivers one of the clearest views yet into what is shared versus truly disease-specific across autoimmunity and why that distinction matters for drug development and patients.
By integrating over 1.3 million immune cells across six autoimmune diseases, the authors move beyond descriptive atlases to identify conserved, mechanistic immune programs that cut across diagnoses, alongside disease-selective features that may explain heterogeneity in outcomes and therapeutic response.
Key findings:
◾ A pan-autoimmune cytotoxic CD8 T cell program marked by clonal expansion and enhanced effector function is observed across multiple diseases, pointing to a shared axis of tissue damage driven by antigen-experienced T cells.
◾ CD14+ monocytes emerge as central orchestrators of humoral autoimmunity, promoting plasma cell survival through elevated TNFSF13B (BAFF) signaling, a pathway already clinically validated but now placed in a broader cross-disease cellular context.
◾ Systematic gene-cluster analysis disentangles cell-intrinsic programs from disease-driven transcriptional states, offering a roadmap for biomarker discovery that is more robust than single-gene approaches.
◾ A machine-learning classifier trained on these single-cell signatures achieves high accuracy in distinguishing autoimmune diseases, highlighting the diagnostic potential of immune-state–based classification rather than symptom-based labels.
This work reinforces a shift underway in immunology and therapeutics: diseases may differ at the organ level, but pathogenic immune circuits are often conserved. Targeting these shared circuits, such as cytotoxic CD8 T cell differentiation or monocyte-plasma cell crosstalk, could enable platform therapies spanning multiple indications, while disease-specific modifiers guide patient stratification and combination strategies.
Equally important, the study highlights how single-cell and AI-enabled frameworks can bridge discovery and clinic, enabling earlier mechanism-based diagnosis, smarter trial design, and clearer go/no-go decisions in immunology pipelines.
The challenge now is translation: validating which of these conserved programs are causal, druggable, and safely modulated in patients. Studies like this provide the systems-level blueprint needed to move autoimmune drug development from reactive suppression toward precise immune reprogramming.
Read the full article here: https://lnkd.in/eJFTRpBt
#AutoimmuneDisease #SingleCellGenomics #Immunology #DrugDevelopment #TranslationalScience
|
|
Scooped by
Gilbert C FAURE
December 19, 2025 4:35 AM
|
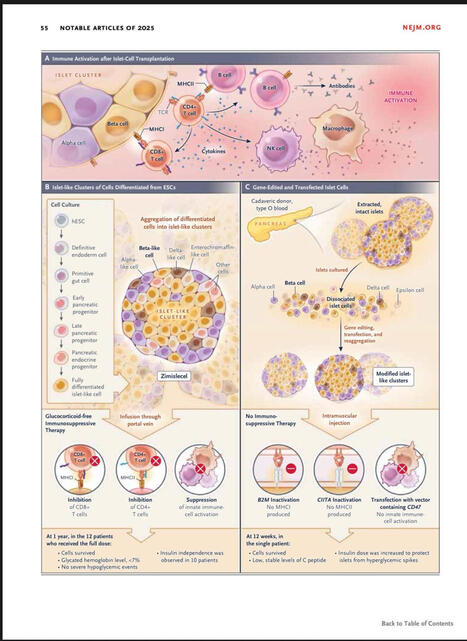
The NEJM has just released the usual end-of-the-year list of most influential papers of 2025.
I found particularly amazing the paper and the editorial concerning allogeneic transplantation of CRISPR genetically-modified Beta cells for type 1 diabetes. A groundbreaking clinical observation that could revolutionise completely this deadly disease. More to come in 2026!
You can download it for free.
|
|
Scooped by
Gilbert C FAURE
December 11, 2025 4:06 AM
|
Autoantibody-triggered podocyte membrane budding drives autoimmune kidney disease
- Chronic kidney disease affects 1 in 10 people worldwide, with damage to specialized blood filter cells of the kidney, called podocytes, playing a critical role.
- In membranous nephropathy (MN), a major cause of nephrotic syndrome, circulating autoantibodies attack proteins on podocyte foot processes (FPs), damaging the kidney’s filtration barrier.
- This study shows that these autoantibodies trigger the formation of antigen-autoantibody aggregates on the podocyte FP plasma membrane.
- These aggregates bud off as stalked vesicles, termed autoimmunoglobulin-triggered extracellular vesicles (AIT-EVs), which are released into the urine.
- AIT-EVs carry disease-causing autoantibodies, their target antigens, essential FP proteins, and disease-associated stressors representing a mechanism for removing immune complexes (ICs) and waste. However, their excessive release leads to podocyte dysfunction and injury.
- The discovery of AIT-EVs bridges the gap between circulating autoantibodies and podocyte responses and can be harnessed in MN patients.
- Clinically, analysis of urinary AIT-EVs represents a novel non-invasive diagnostic tool to sensitively detect and monitor renal autoimmune activity in patients.
https://lnkd.in/ePD3_xDA
#autoimmunity #kidney #kidneydisease #biomarkers
|
|
|
Scooped by
Gilbert C FAURE
July 16, 10:22 AM
|
Macrophage activity is correlated with advanced fibrosis in AILD. To investigate the association between macrophage activation and fibrosis-associated transcriptional programs across AILD, we performed RNA-Seq of cryopreserved tissue samples from 64 clinically indicated liver biopsies of pediatric...
|
|
Scooped by
Gilbert C FAURE
June 22, 10:42 AM
|
Mass spectrometry reveals pneumonia type–specific protein signatures in bronchoalveolar fluid. To identify pneumonia type–specific biomarkers, we sampled BALF and plasma from individuals enrolled in the Successful Clinical Response in Pneumonia Therapy (SCRIPT) study at Northwestern Memorial...
|
|
Scooped by
Gilbert C FAURE
June 11, 1:45 PM
|
|
|
Scooped by
Gilbert C FAURE
May 26, 3:59 AM
|
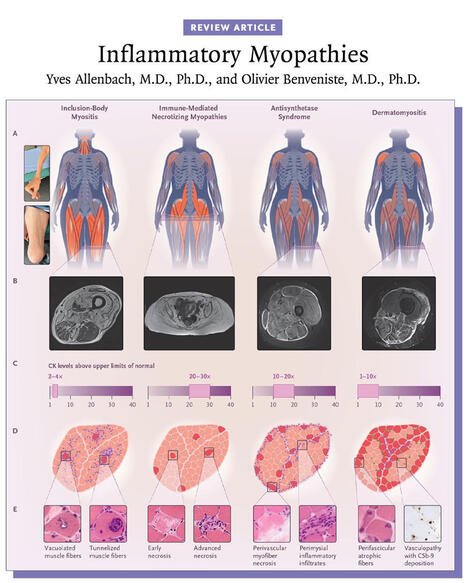
Inflammatory myopathies are typically associated with a myositis-specific autoantibody that determines the diagnosis and prognosis. Myositis subgroups have distinct pathomechanisms that now allow for targeted therapies. 👉 https://nej.md/49Iy9Oo
Inflammatory myopathies are a heterogeneous group of autoimmune diseases characterized by immune-mediated damage to skeletal muscle. They are classified into five major subtypes: inclusion-body myositis, immune-mediated necrotizing myopathies, antisynthetase syndrome, overlapping myositis, and dermatomyositis, each with distinct clinical features and outcomes.
Inclusion-body myositis and immune-mediated necrotizing myopathies primarily affect muscle, with prognosis largely determined by functional impairment, whereas antisynthetase syndrome, overlapping myositis, and dermatomyositis are systemic diseases that can involve the skin, joints, and lungs and may be life-threatening.
The majority of inflammatory myopathies are associated with myositis-specific autoantibodies, which inform diagnosis, subtype classification, and prognosis. Advances in understanding the distinct pathomechanisms underlying each subgroup now enable increasingly targeted therapeutic approaches.
Learn more in the Review Article “Inflammatory Myopathies” by Yves Allenbach, MD, PhD, and Olivier Benveniste, MD, PhD: https://nej.md/49Iy9Oo
#Neurology #Rheumatology
|
|
Scooped by
Gilbert C FAURE
May 13, 3:28 AM
|
Autoantibody flares are important drivers of pathology in systemic lupus erythematosus (SLE), highlighting the pivotal role of B cells in initiating and propagating chronic autoimmunity. Although autoreactive specificities are a normal feature of the naive B cell repertoire, these cells are normally suppressed by layered tolerance checkpoints that limit inappropriate activation. Autoimmune-prone environments can lower these tolerance thresholds, rendering naive autoreactive B cells more sensitive to aberrant cues. Cytokines and other microenvironmental signals shape tissue niches that direct autoreactive B cells towards either germinal-centre or extrafollicular differentiation pathways. Germinal centres support the entry, selection and diversification of autoreactive B cells, with T cell help sustaining these repertoires. By contrast, naive autoreactive B cells entering the extrafollicular pathway exhibit an attenuated requirement for cognate T cell help and strong dependence on complement and TLR signalling. Emerging evidence continues to refine our understanding of germinal-centre and extrafollicular responses as complementary sources of autoreactive effector cells. With this progress, investigations into the origin, development, longevity and tissue dynamics of autoreactive memory B cells as chronic sources of autoantibodies are warranted. Although broad B cell depletion therapies have yielded benefit, a key challenge now is developing precision strategies that selectively target pathogenic B cell subsets. Autoreactive B cells normally held in check by tolerance checkpoints can be driven towards germinal-centre or extrafollicular differentiation in autoimmune environments. This Review examines the cellular, molecular and contextual cues that shape these pathways and considers their implications for systemic lupus erythematosus pathogenesis and therapy.
|
|
Scooped by
Gilbert C FAURE
April 22, 9:47 AM
|
Listen to this episode from Rheumatology Notebooks on Spotify. Podcast on "2023 ACR/EULAR antiphospholipid syndromeclassification criteria".Created with NotebookLM.Intro and outro music created with Suno.Link to the paper: http:// dx.doi. org/ 10. 1136/ ard- 2023-224609
|
|
Scooped by
Gilbert C FAURE
April 10, 3:59 AM
|
New research suggests that amyotrophic lateral sclerosis, or ALS, may be partly an autoimmune disease. Scientists discovered that inflammatory immune cells called CD4+ T cells in people with ALS mistakenly attack a protein found in neurons, known as C9orf72. This self-directed immune response appears to drive rapid disease progression, explaining why ALS can destroy motor neurons quickly and lead to severe physical decline.
The study also revealed two distinct patient groups. One group had highly inflammatory CD4+ T cells and shorter predicted survival, while the second group had more anti-inflammatory CD4+ T cells alongside the harmful ones. These protective T cells appear to regulate the immune response, slowing disease progression and allowing some patients to live significantly longer. Understanding this balance between harmful and protective T cells may help explain why survival in ALS varies dramatically, from just a couple of years to several decades in rare cases.
These findings open the door to potential new treatments that boost protective T cell responses and limit harmful inflammation. Future therapies might harness the immune system to slow ALS progression. The study also has implications for other neurodegenerative diseases, such as Parkinson’s, Huntington’s, and Alzheimer’s, where immune cell involvement may similarly influence disease outcomes.
Research Paper 📄
DOI: 10.1038/s41586-025-09588-6
|
|
Scooped by
Gilbert C FAURE
February 16, 1:00 PM
|
|
|
Scooped by
Gilbert C FAURE
January 15, 4:04 AM
|
Interpretable inflammation landscape of circulating immune cells
- Inflammation represents an altered state within the immune system, which can manifest as either a protective or a pathological response. The cellular and molecular mediators of inflammation play pivotal roles in nearly every human disease.
- Although major progress has been made in characterizing inflammation in specific diseases, a global, holistic understanding is still elusive.
- In this study, the authors leveraged advances in single-cell transcriptomics to delineate inflammatory processes of circulating immune cells during infection, immune-mediated inflammatory diseases and cancer.
- A single-cell atlas of more than 6.5 million peripheral blood mononuclear cells from 1,047 patients (56% female, 43% male) and 19 diseases was generated allowing to learn a comprehensive model of inflammation in circulating immune cells.
- Beyond a disease-centered classification, the authors modeled the expression profiles of inflammatory molecules to uncover discriminative genes related to immune cell activation, migration, cytotoxic responses and antigen presentation activities.
- A classifier framework based on peripheral blood mononuclear cells (PBMCs) was generated, demonstrating the potential of circulating immune cells to contribute to precision medicine strategies for patients suffering from acute or chronic inflammation.
- As limitations of this study:
>Most samples derive from individuals of European ancestry, and expanding to ancestrally diverse populations will be essential to capture global immune variability.
>The classifier requires prospective validation in independent, multicenter cohorts to assess robustness and clinical applicability.
> Understanding the relationship between circulating and tissue-resident immune cells remains key for diagnostic translation.
https://lnkd.in/d7bKEg4T
#immunology #scRNA #omics #autoimmunity
|
|
Scooped by
Gilbert C FAURE
January 12, 6:41 AM
|
More excellent #scicomm from Unbiased Science written by Aimee Pugh Bernard, PhD, Leigh Baxt, and Jess Steier. Learn more about the science behind Lupus and new treatments on the horizon for this autoimmune disease likened to "the wolf within."
|
|
Scooped by
Gilbert C FAURE
December 28, 2025 5:29 AM
|
Recent advancements in the field of biomedical research have unveiled profound insights into Sjögren’s syndrome, a chronic autoimmune disorder primarily affecting the exocrine glands. This condition is notorious for causing significant dry mouth and dry eyes, but its effects extend far beyond...
|
|
Scooped by
Gilbert C FAURE
December 18, 2025 4:42 AM
|
Fatty liver disease is not just a liver problem.
It is a whole-body metabolic disease.
A major review by Prof. Norbert Stefan, Hannele Yki-Järvinen and Brent A. Neuschwander-Tetri shows that metabolic dysfunction-associated steatotic liver disease affects nearly 4 in 10 adults worldwide and most people with it do not die from liver failure, but from heart disease and cancer…
Important:
• Fatty liver comes in different types, driven by metabolism, fat distribution, or genetics
• The liver actively worsens blood sugar, cholesterol, and clotting risk
• Weight loss helps many…but not everyone responds the same way
• Some treatments improve liver health even without weight loss
The key message is simple:
One-size-fits-all medicine does not work for fatty liver disease!!!
Prevention, early detection and treatment need to focus on metabolic health across the whole body, not just liver enzymes…
If we treat fatty liver early, we also reduce diabetes, heart attacks and strokes later 🤞🏼
What should come first in routine care: liver screening, metabolic screening or both?
If useful, save, share or add your perspective below.
Read original review in The Lancet Group: https://lnkd.in/g3m4tA3k
#metabolichealth #fattyliver #cardiovascularhealth #prevention #publichealth
|
|
Scooped by
Gilbert C FAURE
December 9, 2025 4:12 AM
|
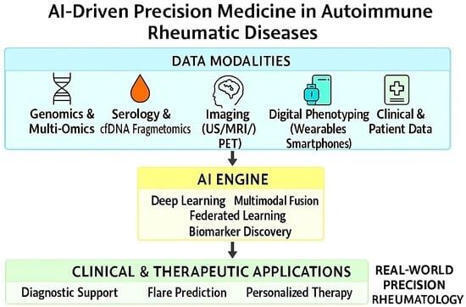
🔥Don't miss this #FeaturePaper by Moawiah Naffaa, PhD and Ola A. Al-Ewaidat.
Emerging AI- and Biomarker-Driven Precision Medicine in Autoimmune Rheumatic Diseases: From Diagnostics to Therapeutic Decision-Making
🔗More details: https://brnw.ch/21wY2dx
#OpenAccess #AutoimmuneRheumaticDiseases #ArtificialIntelligence #Biomarkers #DigitalHealth
|