Your new post is loading...

|
Scooped by
mhryu@live.com
Today, 2:12 PM
|
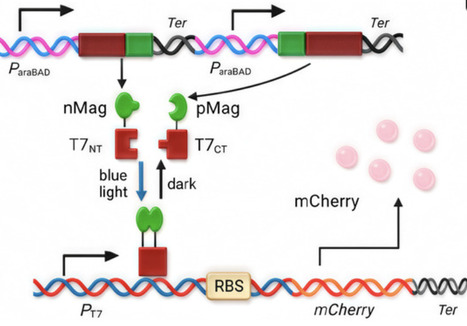
Synthetic biology seeks to build predictable, programmable biological systems. We developed a blue-light-inducible T7RNAP system with dual-input regulation to enable precise spatiotemporal gene control, which is vital for biomanufacturing, therapy, and microbial engineering. We optimized it by replacing RBS sequences, testing tandem T7 promoters, and evaluating split-T7RNAP variants. Expression and bactericidal efficacy were assessed via fluorescent output and real-time growth curves under blue light. RBS variants caused up to 50-fold differences in expression. Three tandem T7 promoters provided the best balance between yield and fidelity. Integration of a benzoate-responsive module enabled 4.5-fold repression at 3 mM benzoate, demonstrating effective chemical off-switching without compromising light induction. This system combines blue light precision with environmental responsiveness, offering non-invasive, on-demand activation for antimicrobial therapy or spatial bioproduction. The benzoate-triggered off-switch is especially valuable for ecological applications such as biocontainment or bioremediation, where gene expression must shut down upon detection of pollutants, for example, aromatic hydrocarbons. Its orthogonal, modular design supports context-dependent control, making it ideal for environmental biosensors, programmable probiotics, and smart antimicrobial delivery in complex ecosystems.
|
|
Scooped by
mhryu@live.com
Today, 1:21 PM
|
Engineered bacteria are emerging as powerful tools in the development of cancer therapies, driven by advances in synthetic biology and tumor immunology. These microbes preferentially colonize solid tumors, where they can deliver therapeutic agents directly to malignant cells and into the tumor microenvironment, inducing tumor cell death and activating robust anti-tumor immune responses. Current strategies include programming bacteria to secrete toxins, tumor-suppressor or pro-apoptotic proteins, and to mediate targeted intracellular delivery. Bacteria can also be engineered to sense tumor-specific metabolites and to adhere to tumor-associated cell surface antigens, further enhancing selectivity and safety. Engineered strains synergize with immunotherapies — including immune checkpoint inhibitors and chimeric antigen receptor-T cells — and can stimulate both innate and adaptive immune responses, even at distant metastatic sites. Here, we review recent progress in this field, with a focus on engineering strategies and their effectiveness in preclinical in vivo tumor models, and outline prospects for future developments and remaining challenges.
|
|
Scooped by
mhryu@live.com
Today, 12:48 PM
|
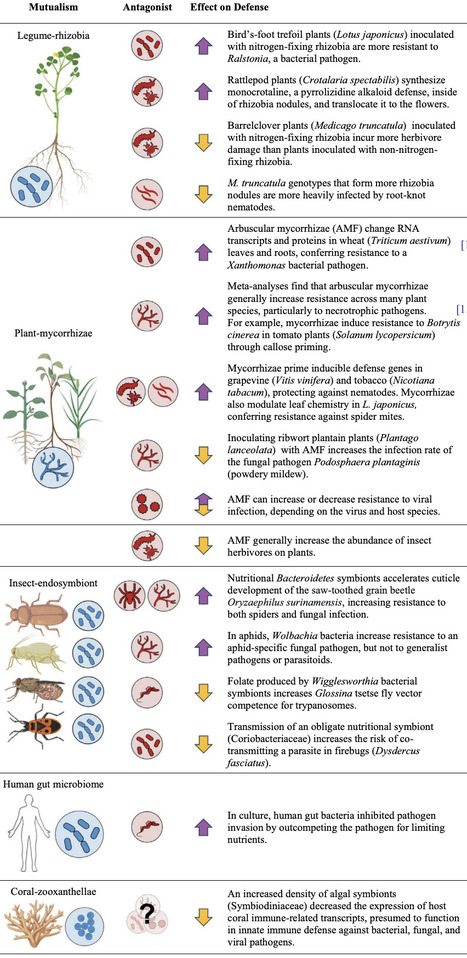
Historically, mutualisms have been categorized by their benefits: nutrition, defense, or transportation. However, many nutritional mutualists have secondary effects on defense. In this review, we propose that nutritional mutualists are major, overlooked drivers of defense evolution and identify four distinct mechanisms by which they can affect the evolution of defense traits. Direct tests of all four mechanisms are scarce. We argue that this is because most work has focused on mutualist effects on trait expression rather than on the parameters that govern evolution: genetic variances, genetic correlations, and natural selection. We highlight new questions that this perspective brings into focus and outline experiments to test them. Finally, we propose that overlap in mutualism and defense resource budgets can unite mutualism and defense theory.
|
|
Scooped by
mhryu@live.com
Today, 11:14 AM
|
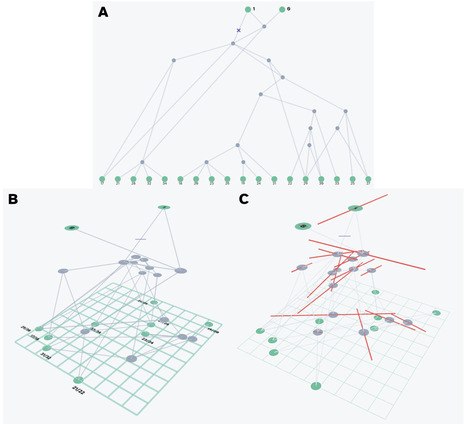
Ancestral recombination graphs (ARGs) are increasingly central to modern population genetics, yet ARG-based methods for spatiotemporal demographic inference remain underutilized in empirical settings due to fragmented workflows and a lack of exploratory tools. ARGscape addresses this by providing a unified framework, seamlessly integrating established and novel tools for ARG simulation, manipulation, and spatiotemporal inference into both graphical and command-line interfaces. ARGscape features dynamic 2- and 3-dimensional visualizations and a novel “spatial diff” visualization for quantitative comparison of ARG-based geographic inference methods. By integrating these various functionalities, ARGscape facilitates novel data exploration and hypothesis generation, bridging the gap between methods development and empirical adoption, and enabling educational uses.
|
|
Scooped by
mhryu@live.com
Today, 1:04 AM
|
Abiotic stress frequency and intensity are increasing, severely impacting plants' health, hence leading to significant crop yield losses (~20%–40% globally). In addition to modifying their genetic and physiological traits to increase stress tolerance, growing research revealed that plant–microbiome interaction plays a remarkable role in determining stress resilience. This review integrates physiological, ecological, and multi-omics data suggesting holobiont plasticity is an unifying paradigm for mechanistic understanding of stress-induced plant–microbe system reorganization. Abiotic stress causes rapid changes in plants' root metabolism and root exudate composition, which alter the release of organic acids, phenolics, osmolytes, and signaling compounds, which selectively change the microbial community's structure in the rhizosphere and endosphere. Microbial taxonomic diversity usually declines under stress conditions. Meanwhile, functional redundancy within the microbial communities is generally maintained or can increase. However, network connectivity may often remain stable or become stronger under stress, and the centrality of keystone taxa usually increases. These keystone microbes play a critical role in sustaining microbial community structure and function. Microbial regulation of phytohormones (such as auxin, ethylene, and abscisic acid), along with control of redox balance, osmotic adjustment, and nutrient cycling, improves plant water use, nutrient uptake, and root development. This often makes them more tolerant to stress by 15%–60% without increasing their biomass. Holobiont plasticity emerges as a quantifiable and potentially predictive characteristic of plant stress responses by integrating microbial network structure, functional gene profiles, metabolomic responsiveness, and host regulatory mechanisms. These responses function on ecological timescales (days to weeks), preceding the more gradual process of host genetic adaptation. This halobiont plasticity-based framework shows promising potential but requires validation under field conditions to prove its robustness and applicability. This opens new avenues for microbiome-assisted plant growth and development of a climate-resilient agricultural system.
|
|
Scooped by
mhryu@live.com
Today, 12:16 AM
|
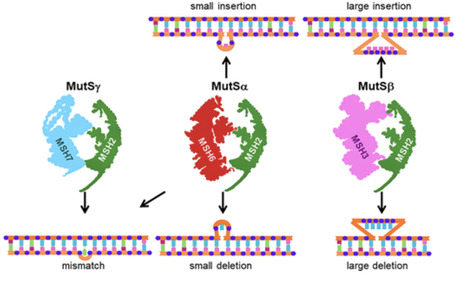
The mismatch repair (MMR) system is an essential DNA repair mechanism that recognizes and corrects single base–base mismatches and unpaired nucleotides that escaped the proofreading exonuclease activity of DNA polymerases or recombination intermediates. This pathway is highly conserved throughout evolution. However, the nature and number of MMR proteins differ between eukaryotes and prokaryotes. Even more, the plant MMR system contains an ancient duplicated MMR protein. In addition, developmental processes vary among eukaryotic organisms. One striking feature is plant genome stability maintenance over multiple generations because embryogenesis and seed development occur after many divisions during plant vegetative growth. Thus, it was of our interest to review the present state of knowledge with respect to the MMR mechanism from eukaryotic organisms, with special comparisons between human, yeast, and plant systems.
|
|
Scooped by
mhryu@live.com
June 25, 11:35 PM
|
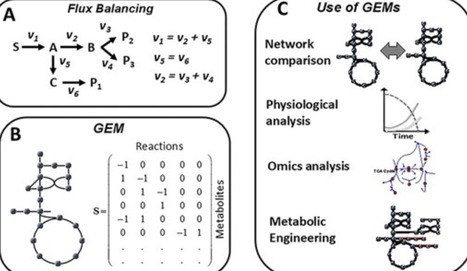
Metabolism underpins cellular function by supplying energy, biosynthetic precursors, and redox balance and in yeast there are thousands of metabolic reactions that are tightly coordinated through multilayered regulation. The yeast Saccharomyces cerevisiae has become a central model for studying metabolism and its regulation and following publication of its genome in 1996, this yeast became pivotal in systems biology. Systems biology integrates experimental data with mathematical modeling to analyze complex cellular networks. A major advance for metabolic analysis was the development of flux balance analysis and genome sequencing enabled reconstruction of the first genome-scale metabolic model (GEM) for yeast. This initial GEM described how hundreds of genes, reactions, and metabolites interact across compartments. Subsequent models, including Yeast8 and Yeast9, expanded the coverage and predictive power, and these models enable metabolic comparison, physiological analysis, omics integration, and design of strains that can be used for production of chemicals and biopharmaceuticals. Overall, S. cerevisiae remains a cornerstone of systems biology and biotechnology, with continued advances expected in integrative modeling and engineering applications.
|
|
Scooped by
mhryu@live.com
June 25, 11:23 PM
|
Microorganisms can cope with stress by entering dormancy, a viable state of reduced metabolic activity that enables persistence, dispersal, and long-term survival. However, microbial life in environmental systems is best understood as a spectrum of metabolic activity, spanning from highly active, dividing cells to deeply dormant phenotypes. This spectrum reflects dynamic survival strategies under fluctuating conditions, with critical implications for ecosystem stability, gene dissemination, and resilience to disturbances in natural and human-influenced systems. Yet, microbial activity is often treated as binary (active vs. dormant), oversimplifying a biological continuity that remains technically difficult to quantify. Here, we synthesize advances in microbial dormancy to reconceptualize activity as a spectrum. We review current and emerging methods to quantify environmental activity, linking each to the Central Dogma of molecular biology (DNA to RNA to protein) to guide interpretation along a generalizable continuum. Through a literature synthesis of terrestrial, aquatic, and wastewater treatment ecosystems, we compare how methods estimate active cells and populations. We recommend standardized reporting of total community size, active cell abundance, and proportional activity to enrich the interpretation of microbiome ‘omics data, with activity intensity and active-inactive switching providing deeper insights. To achieve this, we advocate for increased accessibility and throughput of precise activity-discriminating technologies, alongside renewed use of reliable methods like direct cell counts and activity stains. Adopting this spectrum-based perspective will improve our ability to tackle key societal challenges, such as understanding microbial contributions to ecosystem function under climate change and gene dispersal at human-environment interfaces.
|
|
Scooped by
mhryu@live.com
June 25, 10:47 PM
|
RNA alternative splicing is a fundamental post-transcriptional mechanism whose dysregulation drives various human diseases. Predicting splicing outcomes is therefore a central challenge in precision medicine. This Review traces the evolution of computational approaches from early statistical heuristics to modern artificial intelligence frameworks. We dissect the methodologies that shape predictive performance, including training data scale, output resolution, splicing event quantification and model complexity. Among these factors, quantitative assessment of splicing events and the increasing complexity of model architectures represent particularly critical axes that define both biological interpretability and computational feasibility. We further describe how these models empower translational applications, from annotating variant effects to guiding antisense oligonucleotide development. Nonetheless, persistent challenges remain, including the interpretation of deep-intronic mutations, isoform-level reconstruction and integration of multimodal data. Together, these perspectives define both the progress achieved and the opportunities ahead for splicing prediction in genomics and medicine. Predicting splicing outcomes is a central challenge in precision medicine. This Review traces the evolution of computational approaches from early statistical heuristics to modern artificial intelligence frameworks to characterize splicing events.
|
|
Scooped by
mhryu@live.com
June 25, 4:20 PM
|
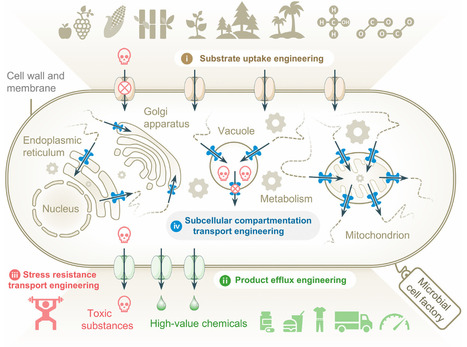
Microbial cell factories are central platforms for sustainable industrial biomanufacturing, yet inefficient transmembrane transport continues to hinder their large-scale industrial translation. Transporter engineering has progressed from trial-and-error overexpression to mechanism-guided rational design, offering effective routes to relieve transport-related metabolic constraints. This review summarizes key advances in substrate uptake, product efflux, stress tolerance, and subcellular compartmentalized transport, analyzes evolutionary trade-offs and practical engineering bottlenecks of transporters, and reviews applications of omics, high-throughput screening, and AI. It aims to deliver a systematic overview of the field and support the further development of transporter engineering for robust industrial microbial cell factories that enable efficient and stable production of high-value chemicals in next-generation green and sustainable biological manufacturing systems.
|
|
Scooped by
mhryu@live.com
June 25, 4:05 PM
|
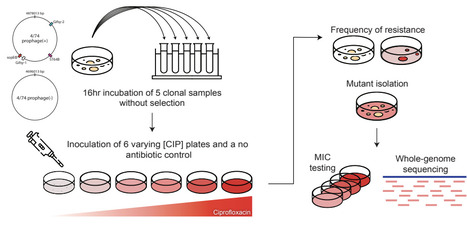
Most naturally occurring bacteria are lysogens, encoding one or more temperate phages integrated into their genome. As prophages are induced by the bacterial SOS response, DNA-damaging antibiotics can trigger SOS-mediated prophage induction, where prophages undergo lytic replication and lyse their host, even at sub-inhibitory concentrations. This prophage-antibiotic synergy therefore sensitizes lysogenic hosts to DNA-damaging antibiotics. However, the mechanism by which prophage-induced sensitization affects the evolution of resistance against these agents is unclear. Here we show that ciprofloxacin-resistant lysogens arise less frequently but exhibit higher levels of resistance following selection. Whole-genome sequencing showed that increased lysogen resistance arose from selection towards mutations in drug targets, efflux pathways, and stress response regulators that reduce antibiotic efficacy or alter SOS induction. Consistent with this result, resistant lysogens exhibited a dampened SOS response, suggesting that prophage induction imposes an additional selective filter on their hosts by eliminating mutants that experience sufficient DNA damage to activate the SOS response. By contrast, prophage carriage had no effect on sensitivity or resistance evolution for antibiotics where DNA damage occurs downstream of the primary mechanism of action. Together, these findings indicate that prophage induction acts as an evolutionary bottleneck that restricts many resistance trajectories while favoring the emergence of rarer, large-effect mutations, potentially accelerating the evolution of high-level resistance.
|
|
Scooped by
mhryu@live.com
June 25, 3:21 PM
|
Bitter taste is a critical quality determinant in food systems, particularly those using sustainable protein hydrolysates, where the unpredictable formation of bitter peptides severely limits consumer acceptance. Achieving predictive control over flavor chemistry requires deciphering the complex sequence-activity relationship. To address this, we integrated the generative capacity of a protein language model with BitterPep-GCN, a Graph Convolutional Network (GCN) capable of robust in silico bitter/non-bitter classification, to target the de novo design of functional bitter and non-bitter sequences. We achieved this by generating two strategic peptide libraries: a targeted tripeptide library derived from known bitter and non-bitter peptide sequences, and a set of de novo designed sequences. For the de novo designed peptides, we fine-tuned the conditional language model ZymCTRL on our curated dataset of sensory-validated bitter peptides (BPS-1000). Both libraries were subjected to classification and rigorous filtering using BitterPep-GCN to select high-confidence candidates for validation. The selected peptides were purchased and rigorously assessed for high purity. Sensory tests were conducted by an expert human panel to determine intrinsic taste quality and taste recognition thresholds. The results validated the high predictive fidelity of our pipeline: out of the 31 tested peptides, 25 were correctly classified, including 15 confirmed bitter and 10 confirmed non-bitter sequences. This study successfully demonstrates the application of machine learning frameworks in the design of bioactive peptides. It provides a set of novel taste-active peptides that can be used to accelerate the rational mitigation of off-tastes in next-generation food products.
|
|
Scooped by
mhryu@live.com
June 25, 3:14 PM
|
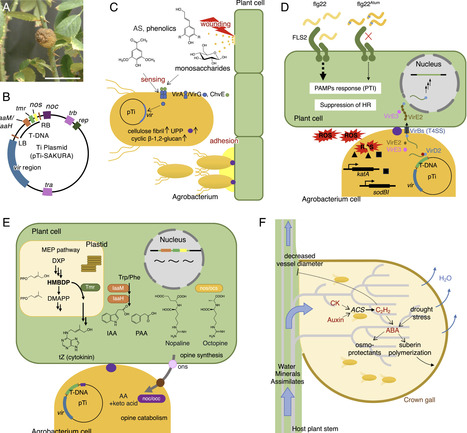
Plants and pathogens engage in complex biochemical communication, mediated by proteins, specialized metabolites, phytohormones and phytohormone-mimicking compounds. These interactions drive a dynamic ‘co-evolutionary arms race’, as plants and microbes compete to gain an advantage, ultimately transforming the infection site into a distinct micro-ecosystem. Microbe-induced galls are abnormal plant organogenesis induced by specific pathogens, creating a niche where the pathogen manipulates the host plant machinery to ensure its own survival. Such galls adversely affect agricultural and horticultural productivity by stunting plant growth and causing deformities. However, the rapid cell proliferation in tumourigenesis, along with the mechanisms driving robust shoot and root re-differentiation, present exciting opportunities for innovative biotechnological applications. Therefore, elucidation of these mechanisms is crucial for advancing basic research with significant potential for agricultural applications. This review focuses on galls induced by Agrobacterium tumefaciens and Rhodococcus fascians—two phytopathogens that utilize phytohormones as tumor-inducing molecules—to highlight the mechanisms underlying plant–pathogen interactions within this specialized microenvironment. It also explores the evolutionary adaptations and strategies of these pathogens. Gaining insight into these biological processes is key to understanding the mechanisms driving biological diversity and evolution, with implications extending beyond plant pathology into the broader field of molecular plant physiology.
|
|
|
Scooped by
mhryu@live.com
Today, 1:27 PM
|
LLPS utilizes dynamic, membrane-less compartmentalization to spatially organize and control biochemical processes, which advances synthetic biology fields such as synthetic metabolic engineering and artificial cell construction, offering novel solutions to longstanding biomedical and biotechnological challenges. However, the rational design and optimization of these promising LLPS-based applications are currently hampered by an incomplete mechanistic understanding of how LLPS precisely governs reaction kinetics. To bridge this gap, we present a comprehensive review that integrates both protein and nonprotein mediated LLPS and systematically dissecting how LLPS orchestrates reaction kinetics─through mechanisms including reactant concentration, reaction–diffusion coupling, microenvironment engineering, and enzyme activity modulation─to dictate bioreaction outcomes. Our analysis begins by outlining the thermodynamic foundations and classifications underpinning LLPS, then critically examines these kinetic regulatory mechanisms, and further summarizes burgeoning applications across biocatalysis, metabolic engineering, diagnostics, therapeutics, origins of life research, and artificial cell construction. Finally, we discuss prevailing challenges and outline strategic pathways for translating LLPS into practical technologies. By synthesizing dispersed knowledge and elucidating fundamental kinetic principles, this review not only fills a critical void in understanding but also establishes essential mechanistic insights and design guidelines to empower the rational development of next-generation LLPS-driven synthetic biology.
|
|
Scooped by
mhryu@live.com
Today, 1:15 PM
|
The gut microbiota is implicated in adverse effects associated with low-calorie sweeteners. Yet, the direct impact of sweeteners on gut bacteria remains largely uncharacterized. Here, we report interactions between 25 phylogenetically diverse gut bacterial strains and 39 commercially used sweeteners. We tested these sweeteners individually and in combination with four commonly co-consumed compounds, viz., advantame, caffeine, vanillin, and duloxetine. Three-quarters of the tested sweeteners individually impacted the growth of at least one tested bacterial strain. Further, over 100 interactions were found between sweeteners and the four co-consumed compounds. Isosteviol, a commonly used sweetener-component, and duloxetine, an antidepressant, synergistically inhibited Roseburia intestinalis, a bacterium previously linked to glucose homeostasis, and Parabacteroides merdae, a prevalent commensal linked to healthy microbiota. Proteomic, metabolomic, and genetic analyses indicate altered small molecule transport underpinning this sweetener–drug synergy. The isosteviol-duloxetine combination also modulated metabolism of a synthetic gut bacterial community, leading to increased toxicity to HeLa cells and altered secretion of inflammation-modulatory cytokines IL-6 and IL-8 by Caco-2 cells. Our data warrant further studies on interactions between low-calorie sweeteners and common xenobiotics.
|
|
Scooped by
mhryu@live.com
Today, 12:41 PM
|
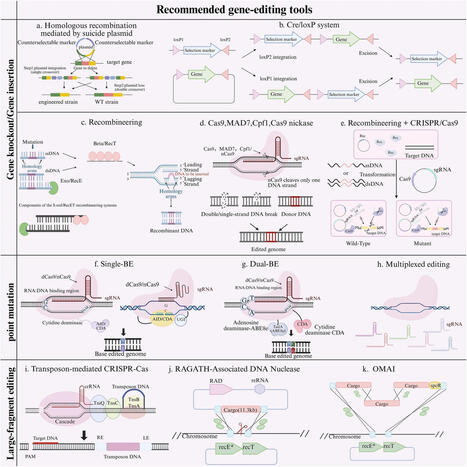
Gram-positive bacteria serve as important chassis microorganisms in synthetic biology, industrial fermentation, and probiotic development. The rapid advancement of gene editing technologies has provided critical technical support for the iterative construction and functional validation of engineered strains. However, due to factors such as cell wall structure, differences in genetic backgrounds, and tool compatibility, the development and editing efficiency of gene editing systems for Gram-positive bacteria still face many challenges. This review focuses on four representative Gram-positive bacterial species-Lactobacillus plantarum, Lactococcus lactis, Bacillus subtilis, and Corynebacterium glutamicum-and traces the evolution and current state of their editing tools, from traditional homologous recombination to CRISPR-Cas9, base editors, and large-fragment integration tools. On this basis, we summarize the common challenges and corresponding strategies concerning host repair capacity, tool compatibility, and inherent limitations of editors in these four bacterial species, and propose recommendations for tool selection based on different application scenarios. This review aims to provide a technical reference for gene editing studies of the above-mentioned bacterial species. Although the conclusions cannot be directly extended to all Gram-positive bacteria, the common issues summarized here may inform the development of gene editing tools for other Gram-positive bacteria.
|
|
Scooped by
mhryu@live.com
Today, 1:19 AM
|
CRISPR-Cas and toxin-antitoxin systems can serve as antiviral defense mechanisms in prokaryotes. In typical toxin-antitoxin systems, toxin activation can limit phage propagation by inducing growth arrest or reduced cellular fitness, while the antitoxin neutralizes toxin activity. Here, we study potential functional synergy between a CRISPR-Cas13a system and a type II toxin-antitoxin module (HicAB) from a Leptotrichia bacterium, when heterologously expressed in E. coli, as well as in biochemical and structural analyses. We show that the antitoxin HicB exhibits toxic properties, and Cas13a directly activates HicB, triggering growth inhibition and conferring protection against bacteriophages. Structural analyses reveal that Cas13a binding promotes the spatial proximity of HicB tetramers, likely enabling its activation. The toxin HicA competitively binds to HicB, thereby inhibiting Cas13a-mediated HicB activation. Importantly, both CRISPR RNA and HicB independently suppress HicA toxicity. Structural evidence indicates that CRISPR RNA forms a hetero-tetradecameric complex with HicAB, occluding HicA’s active site and neutralizing its toxic function. Thus, our findings indicate functional synergy between distinct bacterial immune strategies. CRISPR-Cas and toxin-antitoxin systems can serve as antiviral defense mechanisms in prokaryotes. Here, the authors provide evidence of functional synergy between a CRISPR-Cas13a system and a type II toxin-antitoxin module.
|
|
Scooped by
mhryu@live.com
Today, 12:50 AM
|
E. coli is a key workhorse of biotechnology. Commonly used CRISPR-Cas9 systems for E. coli genome editing are complex and impose metabolic stress on the host, creating demand for more streamlined strategies. Recent studies identified the IS605 transposon-associated TnpB as a programmable RNA-guided (ωRNA) DNA endonuclease, prompting us to explore whether endogenous TnpB in E. coli (EcoTnpB) could be harnessed for genome editing. Biochemical and cellular analyses demonstrated that EcoTnpB efficiently cleaves both chromosomal and plasmid DNA at custom-specified sites in a TAM-dependent manner. Interestingly, E. coli possesses an endogenous recombination machinery capable of repairing EcoTnpB-induced DNA double-strand breaks (DSBs), challenging the long-held view that bacteria lack efficient homologous recombination systems. Based on these findings, we established a single-plasmid editing system (SPEED) in which genome editing is achieved by simply providing ωRNA and a homologous recombination template. By utilizing endogenous EcoTnpB together with the host HR pathway, this system enabled inducible and seamless genome editing at multiple genomic loci in BL21 (DE3), with editing efficiencies ranging from approximately 29% to 56%. Our results demonstrate for the first time that endogenous TnpB can be harnessed for genome editing and may hold potential for broader applications, such as species-specific antimicrobial development.
|
|
Scooped by
mhryu@live.com
Today, 12:15 AM
|
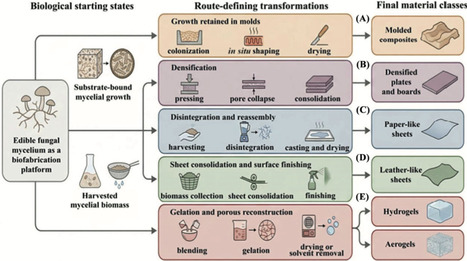
Edible mushroom mycelium-based materials are emerging as renewable, route-dependent systems spanning composites, flexible sheets, hydrogels, aerogels, and hybrid materials, rather than a single engineering class. In this review, we synthesize how strain selection, substrate composition, cultivation conditions, and post-processing shape structure–property–function relationships across these formats. Available evidence is strongest for substrate-bound composites and selected laminate systems, whereas reconstructed gels, aerogels, and functional hybrids remain more exploratory. We assess how synthetic biology, advanced manufacturing, and AI-assisted monitoring may improve controllability and scale-up, while highlighting barriers including reproducibility, moisture sensitivity, durability, techno-economics, and regulatory qualification. Progress will depend on route-specific benchmarking, process control, and validation under realistic service conditions.
|
|
Scooped by
mhryu@live.com
June 25, 11:33 PM
|
Sequence matching algorithms such as BLAST and FASTA have been widely used in searching for evolutionary origin and biological functions of newly discovered nucleic acid and protein sequences. As parts of these search tools, alignment scores and E values are useful indicators of the quality of search results (and the relevance of the matches) from querying a database of annotated sequences, whereby a high alignment score (and inversely a low E value) reflects significant similarity between the query and the subject (target) sequences. For cross-comparison of results from sufficiently different queries, however, the interpretation of alignment score as a similarity measure and E value a dissimilarity measure becomes somewhat nuanced, and prompts herein a judicious distinction of different types of similarity. Via a simulated formulation, we show that an adjustment of E value to account for self-matching of query and subject sequences corrects for certain ostensibly anomalous similarity comparisons, resulting in “regularized” dissimilarity and similarity measures that would be more appropriate for cross-comparisons, as well as database applications, such as all-on-all sequence alignment or selection of diverse subsets. In actual practice, the “regularization” of E value dissimilarity improves clustering and subset selection. While both E value and the “regularized” E value share two of the four axiomatic properties of a metric space, positivity, and symmetry, the latter E value further becomes reflexive and meets the condition of triangle inequality, the remaining two axioms, thus itself an appropriate distance function for metricating protein sequence space.
|
|
Scooped by
mhryu@live.com
June 25, 10:53 PM
|
Microbial communities are shaped by social interactions that influence survival, resource access, and evolutionary trajectories. Most studies of microbial social evolution have focused on freely diffusible molecules such as siderophores and quorum-sensing signals, whereas the physical form in which extracellular products are delivered has received less attention. Bacterial membrane vesicles (BMVs) package enzymes, lipids, nucleic acids, and small metabolites into membrane particles. Membrane packaging slows cargo dispersal, protects labile cargo, and can restrict uptake through receptor-dependent binding. Here, we review vesicle-mediated interactions from the perspective of social evolution, with a primary focus on outer membrane vesicles (OMV) released by Gram-negative bacteria and drawing on Gram-positive examples where informative. We first examine how the route of vesicle biogenesis influences cargo composition and cost to the producing cell, and consider how these differences may influence ecological function. We then discuss how limited dispersal and recipient bias may reduce benefit leakage and thereby help stabilize cooperation. Finally, we consider the conditions under which vesicles support antagonistic outcomes, including toxin delivery, lytic activity, and resource piracy. We argue that whether vesicles favor cooperation, privatization, or competition is not a fixed property but depends on the interplay among biogenesis route, the surface receptor repertoire of recipient cells, local relatedness, and environmental conditions including resource availability and niche overlap. We further highlight that bulk vesicle preparations confound analysis of functionally distinct vesicle subpopulations, underscoring the need to resolve vesicle type, biogenesis route, and recipient identity in spatially structured communities.
|
|
Scooped by
mhryu@live.com
June 25, 4:33 PM
|
Over the past few decades, antibacterial discovery has changed substantially, yet innovation has remained slow despite the growing need for new agents active against multidrug-resistant pathogens. Progress is constrained by scientific hurdles, including target selection, penetration and efflux in bacteria, and the difficulty of translating in vitro activity into in vivo efficacy, as well as persistent economic disincentives. Here, we discuss these challenges and summarize recent advances in the field, with a medicinal chemistry focus on synthetic small molecules that have reached validated-lead or preclinical development and shown in vivo antibacterial efficacy. We also provide an outline of the current clinical pipeline, highlighting areas of innovation and remaining gaps. Our goal is to offer a comprehensive perspective on antibacterial discovery that supports ongoing efforts to strengthen the pipeline in response to antimicrobial resistance.
|
|
Scooped by
mhryu@live.com
June 25, 4:12 PM
|
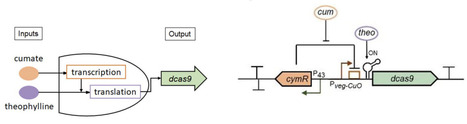
The architecture of Bacillus subtilis biofilms is influenced by the coordinated regulation of cellular specialization, matrix assembly, and metabolism. B. subtilis can form different types of biofilm in diverse physical and chemical environments. Understanding the molecular mechanisms that drive biofilm heterogeneity and adaptation to different environmental niches is crucial for developing more effective strategies to control their formation. In this study, we developed a tightly dual-regulated CRISPR interference (CRISPRi) system and employed multi-scale imaging to investigate the functions of individual genes in two distinct biofilm models: the floating pellicle and the intricate, three-dimensionally structured macrocolony, which develop at the liquid-air and solid-air interfaces, respectively. Our findings validated the CRISPRi approach as a powerful method for studying biofilm development over extended periods and revealed that numerous small non-coding RNAs are involved in regulating biofilm growth dynamics and architecture. The CRISPRi approach was also applied to a pool of 507 genes and transcription units, including protein-coding genes and non-coding RNAs, to screen for cell fitness in these two biofilm models. We discovered that, while both biofilm forms rely on fundamental processes such as cell wall synthesis and nucleotide metabolism, they exhibit different genetic dependencies with regard to matrix composition, motility, and signaling. Exopolysaccharide production, motility, and chemotaxis are crucial for pellicle formation. In contrast, macrocolony development is influenced by γ-polyglutamate synthesis and nutrient acquisition functions. Genes of unknown function were also identified to play a differentially important role in the two biofilm forms. Additionally, the CRISPRi screens revealed further non-coding RNAs regulating biofilm architecture and growth dynamics, adding to the existing layers of post-transcriptional control. Collectively, these results demonstrate that biofilm formation at different physical interfaces is governed by a combination of shared and unique genetic pathways tailored to the specific biofilm environment, thereby opening research avenues into the molecular mechanisms specific to the solid-air and liquid-air interfaces.
|
|
Scooped by
mhryu@live.com
June 25, 3:54 PM
|
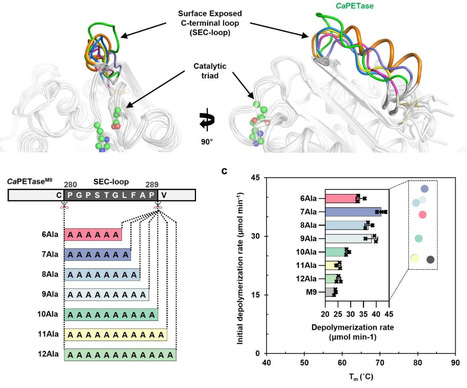
Polyethylene terephthalate (PET) hydrolases have been extensively studied for their potential applications in plastic degradation. However, the structural and mechanistic factors that limit their catalytic efficiency are not yet fully understood. Here, we identify the protruding, surface-exposed C-terminal loop (SEC-loop) in Cryptosporangium aurantiacum PETase (CaPETase) that negatively impacts enzymatic activity by restricting productive access of enzyme to PET. Loop replacement experiments show the non-protruding SEC-loop enhances PET depolymerization rates, despite being ~25 Å from the active site. Kinetic and adsorption studies indicate the non-protruding SEC-loop promotes productive PET access to the enzyme without affecting binding affinity. To further assess the broader applicability of this strategy across diverse PETases, SEC-loop replaced variants of representative PETases are characterized through kinetic and adsorption analyses. We show an engineering strategy focused on modulating enzyme accessibility rather than simply modifying the catalytic site, in rational enzyme design aimed at improving PET degradation efficiency. Polyethylene terephthalate (PET) hydrolases have been extensively studied for their applications in plastic degradation, but the structural and mechanistic factors that limit their catalytic efficiency are not yet fully understood. Here, the authors identify the protruding, surface-exposed C-terminal loop (SEC-loop) in Cryptosporangium aurantiacum PETase that negatively impacts enzymatic activity by restricting productive access of enzyme to PET substrates.
|
|
Scooped by
mhryu@live.com
June 25, 3:16 PM
|
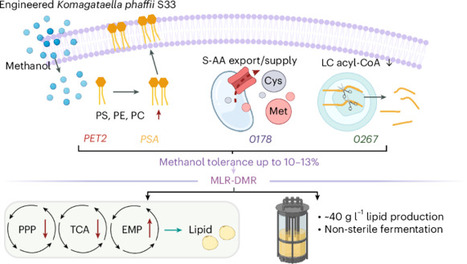
Methanol is a promising C1 feedstock for bioproduction but its cytotoxicity limits applications. Here we enhanced Komagataella phaffii GS115 methanol tolerance to 10–13% (v/v) by overexpressing four genes (phosphatidylethanolamine methyltransferase (PET2), phosphatidylserine synthase (PSA), PAS_chr1-4_0178 (0178) and PAS_chr2-2_0267 (0267)) (S33 strain). Our results show that, under high methanol stress, S33 undergoes membrane lipid remodelling-driven metabolic reprogramming, with central carbon flux being redistributed from the pentose phosphate pathway and citric acid cycle towards glycolysis. Within this framework, 0178 encodes a vacuolar transporter of sulfur-containing amino acids, which we propose supports phospholipid methylation and redox balance. Meanwhile, 0267 encodes a peroxisomal thiolase-degrading long‑chain fatty acyl‑CoAs, limiting toxic long-chain sphingolipid accumulation, maintaining membrane stability. These genes enable S33 to achieve lipid titre of approximately 40 g l−1 under non-sterile fed‑batch fermentation conditions. This work positions S33 as a methanol-tolerant K. phaffii chassis for further strain and process development. Bioproduction from methanol is often challenging owing to the cytotoxicity of methanol. Here a methanol-tolerant Komagataella phaffii chassis is engineered by overexpressing four membrane genes, increasing tolerance to 10–13% methanol and enabling non-sterile lipid production (>40 g l−1) via membrane lipid remodelling and metabolic rewiring.
|