 Your new post is loading...

|
Scooped by
mhryu@live.com
June 19, 4:37 PM
|
Phenotypic biosensors that measure bacterial viability and antimicrobial susceptibility are essential for rapid infectious disease diagnostics, yet their speed is fundamentally limited by the rate at which bacteria encounter reporter molecules, a transport bottleneck that has been typically addressed by complex microfluidic solutions. Here we show that this bottleneck can be overcome by engineering transport directly into the sensing material. A multifunctional ionic hydrogel matrix, co-encapsulating bacterial growth medium and the redox reporter resazurin, exploits swelling-driven convective transport to dramatically accelerate bacteria-reporter interactions without any change to assay chemistry. By systematically tailoring the hydrogel crosslinking density and optimizing the encapsulated nutrient-osmotic microenvironment, we maximize metabolic signal generation to achieve a 12-to-48-fold reduction in detection time relative to solution-phase and conventional hydrogel assays. Deployed in a standard 96-well format for urinary tract infection (UTI) diagnosis of 48 clinical samples, the platform rapidly detects infection in 15 minutes to 2 hours, achieving 95% sensitivity and 100% specificity for bacterial detection, and 100% sensitivity and 98% specificity for antimicrobial susceptibility profiling, compared to time-consuming gold-standard urine culture-based methods. Results are readable both quantitatively on a plate reader and visually as a colorimetric assay, enabling point-of-care deployment without additional instrumentation. Thus, embedding transport enhancement within the sensing matrix, represents a general and scalable design principle for accelerating interaction-limited biosensing, which has excellent scope for rapid diagnostic development.
|
|
Scooped by
mhryu@live.com
June 19, 3:49 PM
|
Methanol oxidation by alcohol oxidases (AOXs) is a key bottleneck in one-carbon (C1) bioconversion due to limited catalytic efficiency and poor formaldehyde tolerance. Here, we report the directed evolution of an alcohol oxidase from Gloeophyllum trabeum, yielding an optimized variant, GtAOXM3. The engineered enzyme exhibits a sixfold increase in catalytic efficiency (7.7 s−1 mM−1), together with enhanced formaldehyde tolerance, thermostability, and high methanol specificity. Molecular dynamics simulations suggest that increased global rigidity and cooperative residue dynamics contribute to the improved performance. When incorporated in multienzyme cascade systems, GtAOXM3 enables efficient conversion of methanol to the value-added chemicals dihydroxyacetone (34.5 mM) and ethylene glycol (23.3 mM). This work establishes GtAOXM3 as an efficient and cost-effective biocatalyst for methanol-based C1 biotransformation.
|
|
Scooped by
mhryu@live.com
June 19, 3:35 PM
|
The immunosuppressive tumor microenvironment (TME), driven by lactate accumulation, critically limits cancer immunotherapy efficacy. In this research article, we engineered Shewanella oneidensis MR-1 by reprogramming energy metabolism to reverse lactate-driven immunosuppression in the TME. Integration of polyphosphate kinase 2 and NAD kinase markedly augmented bacterial bioenergetics, boosting ATP production by 287.5% and elevating the NADH/NAD+ ratio by 299.9% compared with the ‘wild-type’ (WT) strain. This bioenergetic enhancement accelerated targeted lactate depletion and increased intratumoral bacterial persistence, while impairing tumor lactate transport via downregulation of monocarboxylate transporters MCT1 and MCT4. Crucially, metabolic remodeling reversed immunosuppression by enhancing antigen presentation and CD8+ T cell infiltration, and reducing regulatory T cells, thereby converting immunologically ‘cold’ tumors into immunoreactive phenotypes. When combined with anti-programmed cell death protein 1 (αPD-1) immunotherapy, this strategy synergistically amplified antitumor efficacy and established durable immunological memory against recurrence. Collectively, our work establishes a paradigm for bacterial bioenergetic engineering to advance cancer immunotherapy.
|
|
Scooped by
mhryu@live.com
June 19, 3:14 PM
|
Phages, the most abundant biological entities on Earth, infect bacteria and reprogram them into ‘virocells’ with altered physiology and ecology. While metagenomic studies have largely inferred reprogramming through virus-encoded auxiliary metabolic genes (AMGs), phages can reprogram cells through many other tools. In this review, we explore how phage-encoded, host-acting transcription and sigma factors (TSFs) reshape host transcriptional networks beyond simply regulating phage replication. We synthesize emerging genomic evidence for TSF prevalence in phages, mechanistic insights into how host-acting TSFs might influence ecologically relevant cellular functions, and highlight recent experimental and bioinformatic advances that make TSFs particularly tractable for large-scale bioinformatic studies. Together, we position TSFs as AMG-complementary mechanisms of viral reprogramming, tractable for metagenomic inferences, with potential cellular- and ecosystem-level consequences that can power translational applications.
|
|
Scooped by
mhryu@live.com
June 19, 12:18 AM
|
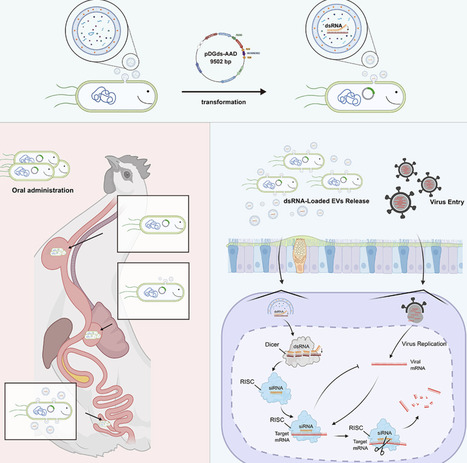
RNA interference (RNAi) is a potent antiviral approach, outperforming traditional pesticides and broad-spectrum drugs. Its use in animal disease control faces two challenges: inefficient target design relying on computer-predicted small-interfering RNAs (siRNAs) rather than virus-derived siRNAs (vsiRNAs), and the lack of cost-effective siRNA delivery systems. In this study, we address both limitations by engineering a probiotic Bacillus subtilis 168 strain, called the recombinant B. subtilis AAD (Anti-AIV-DsRNA, targeted AIV), that constitutively expresses vsiRNA-enriched dsRNA targeting the H9N2 avian influenza virus. Oral administration of AAD leads to the release of double-stranded RNA (dsRNA)-loaded extracellular vesicles (EVs), which efficiently reduce H9N2 viral loads and mitigate pathological lesions. Mechanistically, virus-derived dsRNA is processed by the enzyme Dicer into siRNAs, which then activate RNAi and interferon signaling, resulting in approximately a 70% reduction in viral burden. Overall, these findings demonstrate that integrating the probiotic properties of B. subtilis with EV-mediated dsRNA delivery constitutes a sustainable, effective, and residue-free antiviral strategy for animal disease.
|
|
Scooped by
mhryu@live.com
June 19, 12:06 AM
|
Genome mining now yields tens of thousands of putative biosynthetic gene clusters (BGCs) per project, yet, separating genuinely novel candidates from rediscoveries of known compounds remains the rate-limiting step before experimental validation. Single-axis prioritization tools, antiSMASH similarity, BiG-FAM GCF distance, and self-resistance-enzyme (SRE) filters such as ARTS, each surface a different facet of evidence, yet their isolated use systematically over-ranks rediscovery-prone BGCs and overlooks genuinely orphan clusters. We present novelBGC, a web-hosted framework that converts these disparate outputs into two deliberately non-inverse continuous metrics per BGC, a Novelty (N) and a Reference Similarity (RS) score which together define a 2D decision plane that resolves rediscoveries, divergent family members, contig-edge artefacts, and uncharted chemistry with interactive visualisations, with all component weights user-tuneable at submission. Retrospective validation across three independent experimental datasets demonstrates the utility of the framework for candidate prioritization. Within the first 186-BGC SRE-guided cloning study, every confirmed bioactive product fell within the low-to-mid N band whereas 55 high-N (N ≥ 0.50) BGCs were never selected. Moreover, in the other two studies, it correctly prioritised the fully orphan lariocidin BGC of Paenibacillus sp. M2 and the divergent within-family indanopyrrole-A idp BGC of Streptomyces sp. CNX-425. Together, these case studies demonstrate that the joint (N, RS) space facilitates prioritization decisions that are difficult to achieve using any single criterion alone. from identical input data. novelBGC requires no command-line expertise, no local tool installation, and no manual integration of intermediate output formats, addressing a well-documented accessibility barrier for wet-laboratory researchers engaging with genome-mining workflows. novelBGC is freely available at https://project.iith.ac.in/sharmaglab/novelbgc/.
|
|
Scooped by
mhryu@live.com
June 18, 11:47 PM
|
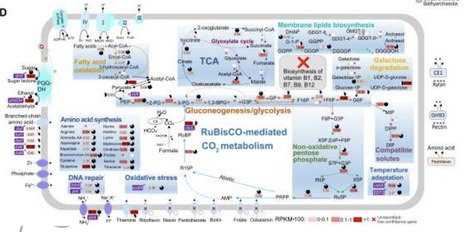
Following the ubiquitous autotrophic ammonia-oxidizing archaea (AOA), heterotrophic representatives of the marine Nitrososphaerota (HMN) form the second most abundant group within this archaeal phylum. However, their eco-evolutionary strategies remain poorly understood. Previous studies have reported a consistent co-occurrence of HMN with marine AOA (MAOA), prompting a detailed investigation into their potential interaction. Through large-scale (meta)genomic and metatranscriptomic analyses, we reveal that HMN possess ultra-streamlined genomes and globally co-occur with marine AOA. The absence of most B vitamin biosynthesis pathways, incomplete citrate cycle and glycolysis, along with the essential requirement for exogenous amino acids, suggest their potential metabolic dependency on AOA. Meanwhile, catalyzed reporter deposition fluorescence in situ hybridization supports a close physical association between HMN and AOA. The nearly synchronous origins of HMN and AOA after oxygen rise, coupled with HMN’s dispersive microhabitats (evidenced by dense, shallow subclades) and extensive horizontal gene transfer between these groups, further support their close relationship—although HMN likely acquired heterotrophic capabilities from bacteria. This study reveals a previously unrecognized association between HMN and AOA, implying a tight coupling between autotrophic and heterotrophic processes in deep-sea habitats.
|
|
Scooped by
mhryu@live.com
June 18, 11:26 PM
|
Glutamine is an important nitrogen donor in the biosynthesis of nucleotides and several other amino acids. Proliferating cells consume high amounts of glutamine, and cell culture media contain glutamine as the most abundant amino acid. Glutamine is industrially manufactured through bacterial fermentation, which requires external supplementation with sugars as the carbon source. Using the cyanobacterium Picosynechococcus sp. PCC 7002, this study aimed to develop a method for the photosynthetic production of glutamine using CO2 as the sole carbon source. The introduction of glutamate dehydrogenase from Corynebacterium glutamicum and glutamine synthase from Saccharomyces cerevisiae increased the concentration of extracellularly released glutamine. Metabolome analysis revealed decreased intracellular citrate levels in glutamine-producing cells. To enhance citrate replenishment, metabolic engineering approaches, including l-lactate assimilation and glycogen deficiency, were examined. The introduction of pyruvate carboxylase and citrate synthase from C. glutamicum significantly increased glutamine production. After optimizing light intensity and CO2 concentration, the recombinant strain produced 1168.5 μM (170.76 mg L−1) glutamine. This study establishes metabolic engineering approaches for converting CO2 into glutamine and demonstrates that cyanobacteria are promising photosynthetic producers of glutamine.
|
|
Scooped by
mhryu@live.com
June 18, 5:01 PM
|
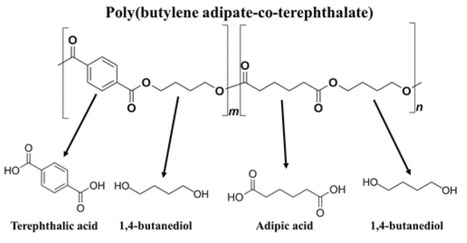
Poly(butylene adipate-co-terephthalate) (PBAT) is an aromatic–aliphatic copolyester widely used in packaging and consumer products. Its aromatic rings confer high resistance to hydrolysis, limiting biological degradation. To enhance PBAT biodegradation, we engineered Paracoccus denitrificans PD1222, a metabolically versatile and genetically tractable bacterium that can accumulate poly(3-hydroxybutyrate) (PHB). A novel plasmid (pV1) was constructed to express the broad-specificity cutinase FsCut under the constitutive Ptuf promoter and fused to the PorG signal peptide for extracellular secretion. Using an optimized transformation protocol, we stably transformed P. denitrificans PD1222 with pV1, enabling secretion of active FsCut and efficient PBAT hydrolysis. In degradation assays, the engineered strain exhibited significantly higher depolymerization rates than the strain carrying the pV0 vector, used as a control. Furthermore, the FsCut-expressing strain accumulated 1.16-fold more PHB than the pV0 strain and exhibited degradation rates of PBAT 2.27-fold higher in enriched medium and 1.9-fold higher in defined mineral medium. These findings demonstrate that targeted expression of a secreted cutinase substantially improves PBAT degradation by P. denitrificans, supporting its potential as a microbial platform for plastic bioremediation.
|
|
Scooped by
mhryu@live.com
June 18, 4:35 PM
|
When E. coli ribosomes are assembled in vitro, manipulation of incubation temperature and magnesium ion concentration has been an essential procedure, which is a crucial step for the assembly of active large subunits. The present study tackles this issue to develop a single-step procedure, which can be performed in near-physiological conditions, where cell-free protein synthesis is active. We found that GTPase factors EngA and ObgE can complement the changes in temperature and magnesium ion concentrations. In the presence of these factors, both the ribosome assembly and translation processes were successfully integrated in the reconstituted cell-free protein synthesis system. Furthermore, we found that these GTPase factors can reassemble the ribosomes to an active state, whose structure was disrupted by EDTA chelation of magnesium ions, indicating that these two factors can reversibly induce the ribosome structure to an intact state. The findings are essential for the bottom-up construction of synthetic cells.
|
|
Scooped by
mhryu@live.com
June 18, 3:56 PM
|
Understanding protein architecture and predicting its structural tolerance to profound remodeling is pivotal for engineering functional proteins. We present SplitSeek-Pro, a deep learning model that evaluates amino acid-level splittability in folded proteins, a property critical for protein engineering tasks such as circular permutation and split reconstitution. By integrating primary sequences with 3D structural features, SplitSeek-Pro achieves residue-resolution predictions through a two-stage training process: large-scale pre-training followed by high-quality fine-tuning. Experimental validation on three distinct proteins confirms its superior predictive power over existing methods. Notably, SplitSeek-Pro identifies characteristic segments that function as cohesive, integral fragments analogous to super-secondary structural motifs. These results establish SplitSeek-Pro as a robust tool for rational protein engineering and offer insights into the fundamental structural building blocks of protein folding. To facilitate community access, we provide an automated web server at http://splitseek.topo.bio. Predicting protein splitability is pivotal for engineering functional variants. Here the authors present SplitSeek-Pro, a deep learning model integrating sequence and 3D features to achieve accurate residue-resolution split site prediction for protein design.
|
|
Scooped by
mhryu@live.com
June 18, 3:20 PM
|
Accurate image annotation is essential for training artificial intelligence (AI) systems in biomedical image analysis, enabling tasks such as cell detection, tissue quantification, and disease characterization. However, creating pixel-level annotations is a time-consuming and labor-intensive process that requires expert input, limiting the development and adoption of AI methods. Recent advances in foundation models, such as the Segment Anything Model (SAM), enable interactive object segmentation from simple user prompts, but their integration into widely used bioimage analysis platforms remains limited and often requires technical expertise. Here we show that SAMJ, a user-friendly plugin for ImageJ/Fiji, enables fast, interactive, and accurate image annotation on standard computers without requiring programming skills or specialized hardware. SAMJ integrates efficient SAM variants into a familiar graphical interface, allowing users to delineate objects in large scientific images in real time using simple clicks or bounding boxes. This approach significantly reduces annotation effort, accelerates dataset creation, and broadens access to advanced AI-assisted annotation tools for the biomedical research community. García-López-de-Haro and colleagues present SAMJ, a plugin that brings the Segment Anything AI to ImageJ/Fiji, enabling fast, click-based image annotation on standard computers, and accelerating creation of biomedical training datasets.
|
|
Scooped by
mhryu@live.com
June 18, 12:43 PM
|
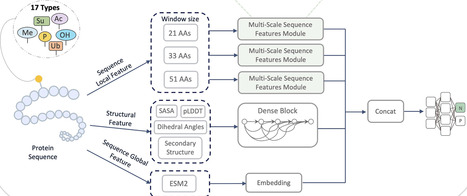
Post-translational modifications are important for regulating cellular functions. Although traditional experimental methods accurately identify PTM sites, they are time-consuming. In this study, we propose a novel model capable of predicting 17 types of PTMs through multi-modal integration and AlphaFold predictions. Our model employs an enhanced CNN-transformer architecture to capture local dependencies within the sequence, while incorporating structural features and evolutionary patterns to effectively capture complex spatial relationships and global contextual dependencies. Through rigorous cross-validation and testing, our model demonstrates exceptional performance, achieving area under the curve scores of 96.5%, 91.6%, 91.0%, and 89.5% for the prediction of hydroxylation, malonylation, O-linked glycosylation, and phosphorylation, respectively. Notably, our model accurately identified known phosphorylation sites on tau and two recently identified residues linked to pre-tangle stages and early Alzheimer’s disease pathology. This work not only deepens the understanding of PTMs but also holds promise for advancing future research in the prediction of PTM sites and functional annotation.
|
|
|
Scooped by
mhryu@live.com
June 19, 3:52 PM
|
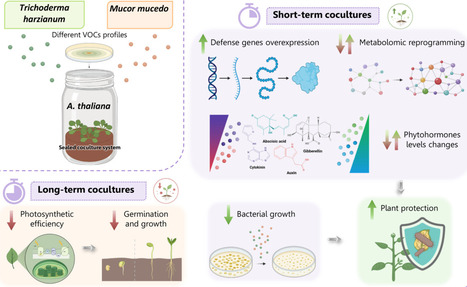
Volatile organic compounds (VOCs) emitted by certain fungi have previously been suggested to modulate plant development and their response to stresses. While some fungal species, such as Trichoderma harzianum, are known to produce VOCs, the production of VOCs in other phylogenetically divergent species, such as Mucor mucedo, was previously unknown. This study aimed to analyze the VOCs profiles emitted by these fungal species and examine their effects on the growth and defensive responses of Arabidopsis thaliana. Volatilome analysis revealed that the most abundant VOCs for both species were alcohols, ketones and esters. However, notable differences were also observed, particularly with regard to terpenes. We then observed that prolonged exposure to VOCs emitted by both fungal species had detrimental effects on A. thaliana growth and photosynthetic performance, whereas shorter exposures enhanced the expression of defense-related genes and the plant defense against the phytopathogenic bacterium Pseudomonas syringae. This increased resistance does not appear to be mediated by canonical H2O2-induced immunity, but rather by subsequent responses that trigger complex metabolic reprogramming, including glucosinolate biosynthesis. Therefore, our results confirm that VOC-mediated plant-fungus interactions are very relevant to plant fitness, highlighting the need to understand them in greater depth.
|
|
Scooped by
mhryu@live.com
June 19, 3:45 PM
|
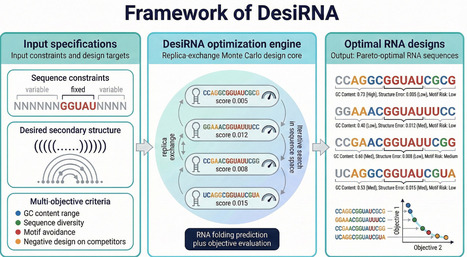
Programs and computational frameworks for predicting RNA sequences with desired folding properties are continually being developed and expanded. A decade has passed since they were last reviewed in this journal, and this brief review provides an update to the review published at that time. Given a target secondary structure, these programs aim to predict RNA sequences that fold into the desired structure while satisfying various constraints. This procedure is known as inverse RNA folding. Traditionally, inverse RNA folding has been used to design optimized RNAs with favorable properties. This updated review covers some of the most widely used freeware programs developed for this purpose over the past decade. RNAinverse, part of the Vienna RNA package, was the first program devised to address the inverse RNA folding problem, and many subsequent programs were described in the earlier review. Some of the most important computational frameworks are the Infrared framework and DesiRNA. In addition, RNA design capabilities have been incorporated into the RNAstructure package, while NUPACK, as well as MoiRNAiFold, MODENA, incaRNAfbinv, and related tools have undergone recent updates. A variety of strategies have also emerged to address the problem of 3D RNA design and RNA–RNA interactions. The various programs mentioned employ distinct approaches, ranging from replica exchange Monte Carlo to constraint satisfaction, as well as Boltzmann sampling and machine learning approaches. Machine learning methods are being developed for emerging applications in biotechnology such as messenger RNA(mRNA) design and CRISPR guide RNA (gRNA) design. This brief review examines these programs and provides a timely update.
|
|
Scooped by
mhryu@live.com
June 19, 3:29 PM
|
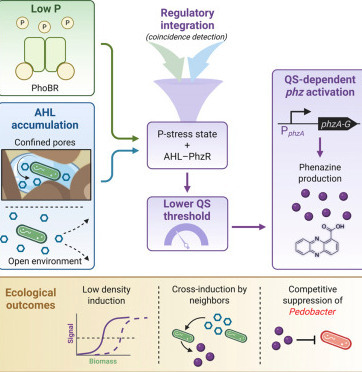
Bacteria often coordinate collective behaviors such as biofilm formation and secondary metabolite production through quorum sensing (QS), a regulatory system traditionally linked to high cell density. However, in environments such as soil, where microbial populations are spatially fragmented, sparse, nutrient-limited, and subject to mass transport, the mechanisms that enable QS-dependent processes remain incompletely understood. Here, we investigate the regulation of a secreted redox-active metabolite, phenazine-1-carboxylic acid (PCA), in Pseudomonas synxantha 2-79, a model rhizobacterium, under phosphorus (P) limitation, a persistent stress in many soils. Using a combination of microscopy and molecular genetic approaches, we show that P limitation sensitizes the QS activation threshold by an order of magnitude, enabling phenazine induction at relatively low population densities compared with P-replete conditions. This induction is abolished in QS-deficient mutants and restored by the addition of exogenous acyl-homoserine lactone (AHL), demonstrating that QS remains essential, but its threshold becomes environmentally tuned. Under P limitation, spatial confinement and pore saturation levels further shape the timing and location of induction, illustrating how physical structure and nutrient stress can modulate bacterial activities. Moreover, P stress confers both collaborative and competitive advantages, enabling P. synxantha to undergo low-cell-density AHL cross-induction with related Pseudomonas spp. and to suppress other rhizobacteria. Lastly, on plant roots, phenazine biosynthetic genes are more strongly induced under P limitation. These findings illustrate how the nutrient status of an environment can modulate the onset of QS, enabling quorum-regulated behaviors to activate at lower thresholds.
|
|
Scooped by
mhryu@live.com
June 19, 3:02 PM
|
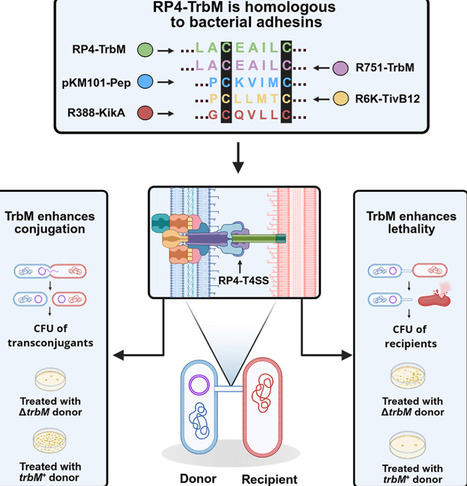
Bacterial conjugation drives horizontal gene transfer and antibiotic resistance via type IV secretion systems (T4SS) on conjugative plasmids like RP4. Previously, we found that the RP4-T4SS mediates interbacterial killing, a process enhanced by the uncharacterized gene trbM. Here, we characterize RP4-TrbM and identify structural features essential for boosting both conjugation and killing. Computational analyses reveal that the RP4-TrbM shares similarities with known bacterial adhesins in other conjugative systems. Homologs from plasmids R751, R388, and pKM101 could complement RP4-TrbM-knockout strains, restoring and enhancing conjugation (R751-TrbM and pKM101-Pep) and conjugation-associated killing (R751-TrbM, R388-KikA, and pKM101-Pep), while TivB12 from conjugative plasmid R6K could not complement the RP4-T4SS. Furthermore, we identified an essential functional domain in RP4-TrbM that retains activity even when repositioned between a different signal peptide and C terminus. These findings expand our understanding of the RP4 conjugative machinery and highlight TrbM-like proteins as promising targets for inhibiting T4SS-mediated processes.
|
|
Scooped by
mhryu@live.com
June 19, 12:10 AM
|
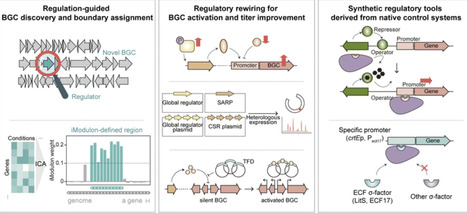
Streptomyces species are well known for their immense genomic potential for the discovery of natural products. However, the majority of their biosynthetic gene clusters (BGCs) remain silent or are poorly expressed under laboratory conditions. This silence is largely attributed to the regulatory complexity encoded within their large genomes, which feature thousands of regulators and multilayered control systems. In this review, we summarize the current knowledge regarding genome-scale transcriptional regulation in Streptomyces, alongside the emerging experimental platforms designed to investigate these mechanisms. By integrating and comparing fragmented data on regulatory architectures, we highlight the extensive hierarchical, combinatorial, and condition-dependent regulatory networks that govern secondary metabolite biosynthesis in Streptomyces. Furthermore, integrative analyses reveal the conservation of regulatory architectures across Streptomyces species, facilitating the translation of findings from model organisms to BGC discovery, activation, and engineering in less studied species. Beyond transcription, we also discuss the additional regulatory layers, including post-transcriptional, translational, post-translational, and chromosome topology-based controls, and their practical applications in natural product research. Collectively, this review reframes the complex transcriptional regulatory networks not as a bottleneck but as a central principle for understanding and exploiting the biosynthetic potential of Streptomyces.
|
|
Scooped by
mhryu@live.com
June 18, 11:58 PM
|
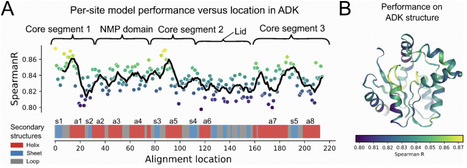
Temperature is a fundamental determinant of bacterial physiology and ecology. Optimal growth temperature (OGT) is highly variable across species, contributing to differences in where and when species are most likely to thrive. Although the OGTs for most bacteria remain unknown, the increasing availability of genomes from uncultivated and cultivated taxa has made it advantageous to build genomic, cultivation-independent models to infer OGT. However, pre-existing genomic models often lack the generalizability and mechanistic grounding required for robust inferences of OGT. We propose a novel framework for predicting bacterial OGT which uses learned protein structural signatures of thermal adaptation. We hypothesize that biophysical tradeoffs which dictate enzymatic functions across variable temperatures provide a more robust empirical basis for OGT prediction than broad genomic features. Our OGT-predicting model, ROSEATE, is based on a single gene, adenylate kinase (ADK), that encodes for a ubiquitous enzyme essential for energy homeostasis. ROSEATE uses high-dimensional latent space encoding via MSA Transformer, a protein language model which embeds ADKs in a manner which preserves biophysical information about embedded proteins. We show that the accuracy of the ROSEATE model is on par with other genome-based models, has a high degree of phylogenetic generalizability, and the ESM embeddings effectively capture key temperature-adaptive enzyme characteristics derived from AlphaFold structures. Because ROSEATE is based on analyses of a single ubiquitous protein, it can be used with metagenomic data to infer the community-level variation in bacterial OGTs. We demonstrate this feature of ROSEATE by reconstructing ADK sequences from over 500 environmental and host-associated metagenomes, successfully distinguishing community-wide thermal preferences across diverse habitats, from polar oceans to mammalian guts. By transitioning from genomic proxies to informationally dense protein structural features, this work provides an efficient, interpretable tool for predicting bacterial OGTs across taxa and whole communities.
|
|
Scooped by
mhryu@live.com
June 18, 11:42 PM
|
Acidobacteriota, one of the most abundant and ubiquitous bacterial phyla in soils, are well recognized for their role in carbon cycling. In contrast, their roles in soil nitrogen cycling remain largely unexplored, although recent metagenome-assembled genome (MAG) analyses suggest that Acidobacteriota may harbor genes involved in nitrogen cycling. Here, we provide culture-based evidence of diazotrophy within this phylum and demonstrate the widespread occurrence of nitrogen-fixing Acidobacteriota across diverse soil types. From grassland and agricultural soils, we isolated five Acidobacteriota strains representing novel taxonomic lineages, four of which harbor functional nitrogenase (nif) gene clusters. These strains were capable of fixing atmospheric nitrogen in vitro and/or in soil microcosms, as evidenced by acetylene reduction, N2-dependent growth, transcription of nif genes, incorporation of 15N into biomass and soil, and inhibition of nitrogenase activity by ammonium. Furthermore, global-scale meta-analysis of soil metagenomes revealed that nif-harboring Acidobacteriota are widely distributed and locally dominant across soil types. These results demonstrate the nitrogen-fixing capability of Acidobacteriota at the organismal level, complementing MAG-based inferences, and underscore their adaptive capacity in nitrogen-limited environments and their potential contribution to terrestrial nitrogen fixation. We also propose novel taxa within the class Terriglobia of the phylum Acidobacteriota, including diazotrophic strains, comprising one novel family, three novel genera, and four novel species: Koromonadaceae fam. nov., Koromonas soli gen. nov., sp. nov., Koromonas humicola sp. nov., Oryzophilus luti gen. nov., sp. nov., and Humiphilus diazotrophicus gen. nov., sp. nov.
|
|
Scooped by
mhryu@live.com
June 18, 5:07 PM
|
When growing bacteria start to reach stationary phase, the nucleotide guanosine tetra-phosphate (ppGpp) accumulates intracellularly and regulates the transition of cells from growth to growth arrest. Because commonly studied bacteria remain viable in stationary phase only briefly under laboratory conditions, the role of ppGpp in sustaining long-term bacterial survival after growth arrest has not been widely studied. Rhodopseudomonas palustris strain CGA009 is a phototrophic alpha-proteobacterium that survives under anaerobic conditions for months when not growing due to carbon starvation if provided light. When we quantified intracellular ppGpp in growing and growth-arrested R. palustris, we found that it was undetectable in growing cells but accumulated to about 100 µM when cells ran out of carbon and entered the stationary phase. These elevated levels of ppGpp were maintained over a 60-day period of growth arrest. Intracellular GTP was 100–200 µM in growth-arrested cells, and ATP was at 2–4 mM. ppGpp had global effects on gene expression, with over half of the genes in the R. palustris genome being activated or repressed by ppGpp in stationary phase cells. These results suggest that, in addition to its known role in facilitating the transition of bacteria from growth to stationary phase and accompanying stress responses, ppGpp is important for prolonging bacterial survival in stationary phase.
|
|
Scooped by
mhryu@live.com
June 18, 4:55 PM
|
Precise and orthogonal regulation of genetic circuits is a central challenge in synthetic biology, particularly at the translational level where tools remain scarce. Here, we address this by engineering suppressor transfer RNAs (sup-tRNAs) charged with canonical amino acids to enable programmable nonsense mutation readthrough in E. coli. Screening of 20 variants revealed a clear sup-tRNA design rule: readthrough efficiency is dictated by the similarity of the native tRNA anticodon to amber codon (CUA), as it preserves native aaRS interactions. We then demonstrate the power of this tool for advanced genetic circuit engineering. First, in a LacI-based biosensor, sup-tRNA regulation reduced background leakage by >77% and increased the induction dynamic range by 4.3-fold (from 6.67 to 28.68). Second, by dynamically balancing glycolytic flux through targeted pykA and pykF regulation, we increased the titer of N-acetylneuraminic acid by 66% (from 5.33 to 8.82 g/l) without compromising cell growth. Our work establishes engineered cAA-charged sup-tRNAs as a versatile, efficient, and cost-effective platform for precision translational control within genetic circuits, opening new avenues for biosensor optimization and metabolic engineering.
|
|
Scooped by
mhryu@live.com
June 18, 4:14 PM
|
Ancestral sequence reconstruction (ASR) resurrects proteins that existed millions of years ago. These ancient enzymes often display capabilities that modern proteins lack. In this Comment, we explore key ASR applications and future challenges, and showcase how ancient enzymes are inspiring new innovations in biotechnology. Ancestral sequence reconstruction is used to resurrect extinct proteins, which often have capabilities that modern proteins lack and thus applications in biotechnology.
|
|
Scooped by
mhryu@live.com
June 18, 3:47 PM
|
Advancing the performance of programmable genome editing nucleases remains a key challenge in expanding their research and therapeutic applications. Here, we introduce a scalable deep learning–guided protein engineering framework for improving nuclease activity without requiring experimental training data. As a demonstration, we apply this strategy to SpuFz1, a compact Fanzor nuclease of eukaryotic origin, identifying and validating beneficial mutations that produces a multi-mutant variant with an 11.6-fold increase in editing efficiency. In parallel, we use comparative sequence analysis to design and experimentally validate a 75-nt ultrashort ωRNA scaffold, reducing guide RNA length by 79% while maintaining activity. Integration of these optimized components yields enFanzor, a compact genome editing system that achieves editing efficiencies up to 81.9% in mammalian cells, with strong editing performance in both human hematopoietic stem and progenitor cells (HSPCs) and mouse embryos. The outperforming variant developed through this strategy also supports robust CBE and ABE activity. Notably, the shortened ωRNA not only improves nuclease editing specificity but also leads to a substantial increase in base editing efficiency. Together, this work demonstrates the power of combining AI-guided protein optimization with rational RNA design, and establishes a generalizable strategy for engineering next-generation genome editing tools. How to quickly and systematically advance the activity of programmable RNA-guided endonucleases remains a key challenge to be overcome. Here the authors combined deep learning-guided protein optimization with rational RNA design, generating enFanzor system which supports robust genome editing.
|
|
Scooped by
mhryu@live.com
June 18, 12:48 PM
|
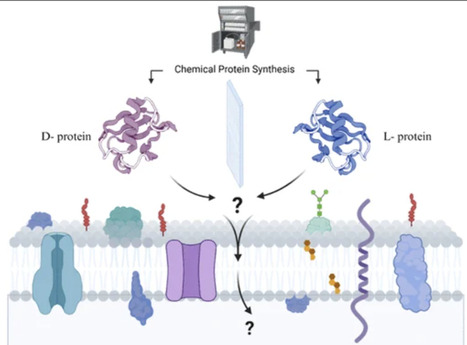
Mirror-image peptides and proteins are attracting interest as therapeutics, as key building blocks for constructing mirror-image life, and as tools to probe the origin of life. Their resistance to proteolytic degradation and unique stereochemistry make D-peptides/proteins particularly appealing for biomedical applications, yet a critical unresolved question is how their intracellular uptake compares to that of natural L-forms. To address this, we systematically investigated the role of cargo chirality in cellular internalization while maintaining a constant delivery vehicle. Three model cargos of increasing size and structural complexity were synthesized in both L- and D-configurations and conjugated to an identical cyclic deca-arginine (cR10) cell-penetrating peptide (CPP). By keeping the CPP scaffold constant, we reduced delivery-related variability and directly assessed the influence of the cargo chirality on uptake. Quantitative uptake analysis using flow cytometry, gel analysis, and confocal microscopy across multiple mammalian cell lines reveals that L-cargos are internalized more efficiently than their mirror image D-counterparts, demonstrating that cargo chirality is a key determinant of uptake efficiency across the chiral biological membrane. Collectively, these findings provide a systematic basis for further exploration of chirality effects in CPP-mediated delivery and may inform the design of mirror image peptides and proteins for therapeutic or synthetic biology applications.
|






















NaAMPS hydrogel is synthesized and then soaked for 10-12 hours in a solution containing M2 growth medium + resazurin (1 µg/mL). Clinical sample (20-100 µL) is pipetted directly onto the hydrogel. Incubate at 37°C. Read fluorescence (560 nm excitation / 610 nm emission) or observe colorimetric shift (purple → pink).