Your new post is loading...

|
Scooped by
mhryu@live.com
Today, 5:26 PM
|
Resource competition theory typically assumes static traits and continuous supply of resources. Yet microbial communities often experience feast-famine cycles and rapid trait change. To investigate coexistence under these nonequilibrium conditions, we integrate modern coexistence theory (MCT) with a genome-scale metabolic model that explicitly links resource use (traits) to metabolic fluxes and growth. MCT partitions competitive interactions into niche and fitness differences, to predict when trait-driven departures from neutrality result in coexistence or exclusion. Using dynamic flux balance analysis, we define a function that maps trait-resource matching to niche and fitness differences between species in a two-species two-resource system. This mapping shows that niche and fitness differences are not independently tunable under resource competition: changes in transporter-mediated resource uptake and changes in resource concentration ratios generate constrained trajectories through coexistence space. Specifically, we show that the minimum niche difference required for coexistence increases linearly with the absolute difference in maximal growth rates on limiting resources, showing how limiting similarity between species can emerge from intracellular metabolic constraints. Furthermore, we find that in batch culture simulations, initial conditions (inoculum size, total resource concentration) determine the timescale of the transient growth phase, with niche differences saturating and fitness differences increasing as the timescale grows, thereby governing competition outcomes. Finally, we test these predictions experimentally using E. coli strains with targeted resource transporter knockouts under both equal and skewed resource concentrations. Our results confirm that transporter-mediated trait changes and resource concentration ratio modulation can be harnessed to engineer coexistence. Together, our work demonstrates that trait-resource matching imposes structured constraints on the joint evolution of niche and fitness differences, thereby shaping biodiversity maintenance in microbial communities under nonequilibrium conditions.
|
|
Scooped by
mhryu@live.com
Today, 5:18 PM
|
The influence of plate tectonics on microbial distributions remains poorly understood. Here, we demonstrate that the global biogeography of hydrothermal vent-endemic chemoautotrophic microbiota is structured by the tectonic history of the global major oceans. These microbiota, particularly anaerobic ones, are significantly more abundant in early-origin Pacific, Arctic, and Mediterranean oceans, whereas they are notably scarce in late-formed Atlantic and Indian Oceans. This pattern was attributed to the timing of ocean formation and its interplay with global redox evolution. Fully oxygenated conditions in the Phanerozoic during the formation of the latter two oceans imposed a dual-dispersal barrier among oceans: toxicity of molecular oxygen to anaerobes and depletion of energy sources (especially reduced chemicals) for aerobes, but such a barrier didn’t exist before the Phanerozoic, when the former three oceans started. These results integrate microbial biogeography into a geodynamic framework, revealing that even microbial life is subject to planetary-scale geological constraints. The links between deep-time geologic processes and microbial distribution remain poorly understood. This paper reveals that tectonic and geochemical evolution shapes the global biogeography of hydrothermal bacteria by limiting their dispersal to the oceans formed after oxygenation.
|
|
Scooped by
mhryu@live.com
Today, 5:05 PM
|
Biotic-abiotic interfaced configurations hold great promise for application in renewable energy and artificial photosynthesis systems. Recent advances in synthetic biology, computational, and visualization techniques, along with enhanced high-resolution characterization, have enabled a deeper fundamental understanding of the interface, which, in turn, has improved electron transfer processes and the design architecture. These developed configurations open new routes to mimic the photosynthetic apparatus or add new applications based on biotic and abiotic catalytic reactions. Aiming to surpass natural systems, researchers have examined methods to reconfigure these block sets into new designs. This review focuses on the advances in artificial photosynthesis and coupled biotic-abiotic biohybrid systems. The work presents the development of artificial photosynthesis configurations aimed at generating light-induced energy or fuels. The use of natural photosynthetic proteins, inorganic photocatalysts, and advanced biohybrid materials is presented and discussed, aiming to enable future biotic-abiotic design and the ambitious goal of developing real-world applications.
|
|
Scooped by
mhryu@live.com
Today, 4:53 PM
|
Katsuya Fuchino works in the field of bacterial cell biology and applied microbiology. In this mSphere of Influence article, he reflects on how the studies “Dependency on medium and temperature of cell size and chemical composition during balanced growth of Salmonella typhimurium” by Schaechter et al. and “A metabolic sensor governing cell size in bacteria” by Weart et al. inspired the field of bacterial cell size homeostasis and his current research. Additionally, he highlights the value of reading classic, original papers that influenced the field, as a means to reflect on paradigm shifts and generate new ideas.
|
|
Scooped by
mhryu@live.com
Today, 4:25 PM
|
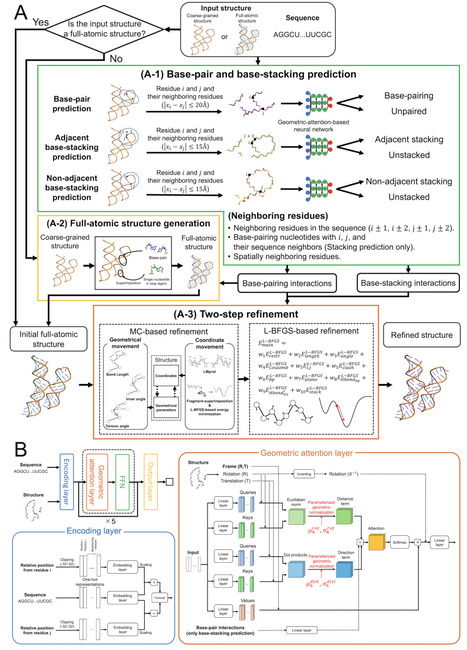
Considerable progress has been made in AI-driven RNA structure prediction, but the resulting models often lack complete atomic details or suffer from severe stereochemical distortions and incorrect local interactions. We present RNArefine, an AI-guided hierarchical framework for atomic-level RNA structure refinement. RNArefine first predicts base-pairing and base-stacking interactions using geometric attention networks and then integrates the interactions with physics-based force fields to guide a two-step refinement strategy consisting of Monte Carlo conformational sampling followed by L-BFGS energy optimization. Large-scale benchmark experiments on both sequence-based prediction models and cryo-EM-derived structures demonstrated that RNArefine consistently improves stereochemical quality, interaction fidelity and physically penalized structural accuracy while preserving global topology. When applied to blind CASP16 RNA prediction models, RNArefine improved ranking scores for 28 of the top 30 groups. These results establish RNArefine as a robust open-source framework for transforming raw RNA folds into physically realistic atomic models for downstream structural and therapeutic applications.
|
|
Scooped by
mhryu@live.com
Today, 4:15 PM
|
Orthogonal gene expression systems based on bacteriophage-derived T7 RNA polymerase (T7 RNAP) offer a promising strategy for decoupling engineered transcription from host regulatory networks. Although recent advances have enabled productive T7 RNAP-mediated expression in Saccharomyces cerevisiae, strategies for independently controlling multiple genes within a shared T7 transcriptional framework remain limited. Here, we developed a modular pYTK-compatible T7 RNAP expression architecture that separates overall transcriptional capacity from gene-specific control of protein output. We established interchangeable T7 promoter, terminator, polyadenylation signal, and 5’ untranslated region (UTR) parts for combinatorial assembly. Analysis of expression cassette dosage showed that transcript abundance and expression output scale with cassette copy number, with high-copy constructs producing transcript levels comparable to those driven by the GAL1 promoter. To enable gene-specific tuning, we constructed a library of fourteen 18 nt UTR elements that generated a ∼38-fold range of protein output from a common T7 promoter. Application of this library to a three-gene β-carotene biosynthesis pathway enabled ∼20-fold variation in product titer through UTR assignment alone, without altering promoter identity or gene copy number. Together, these results establish a modular T7 RNAP expression framework in which cassette dosage defines overall transcriptional capacity while interchangeable UTR elements enable gene-specific tuning of expression output, providing a practical strategy for multigene expression control in yeast.
|
|
Scooped by
mhryu@live.com
Today, 3:32 PM
|
Circular RNA (circRNA) exhibits extended stability and enhanced protein expression, and its translational efficiency and immunogenicity can be improved through nucleoside modification and rolling circle translation (RCT). However, it remains challenging to produce protein-encoding circRNA with modified nucleosides. Here we identified a cap-independent translation enhancer (CITE) element from black beetle virus, termed BBV, which could drive the translation of nucleoside-modified RNA and RCT of engineered circRNA. In addition, we developed a system to produce ‘scarless’ circRNA via in vitro transcription (IVT). The resulting circRNA vaccine outperformed m1ψ-mRNA vaccine in inhibiting tumor growth in mice. With nucleoside modification, circRNA exhibited reduced expression of pro-inflammatory cytokines. Further, nucleoside-modified circRNA encoding glucagon-like peptide-1 (GLP-1) peptides reduced blood glucose levels with efficacy comparable to that of commercial semaglutide, and ameliorated liver damage in obese mice. Moreover, nucleoside-modified circRNA encoding myelin oligodendrocyte glycoprotein (MOG35–55) peptides induced immune tolerance and alleviated disease progression in an experimental autoimmune encephalomyelitis (EAE) murine model. Collectively, we established a nucleoside-modified circRNA platform that facilitated efficient translation with minimal immunogenicity, expanding the application scenarios of circRNA beyond vaccines. A suite of tools to improve translation and immunogenicity of circular RNAs, including nucleoside modification and rolling circle translation, for applications in cancer vaccines, protein replacement and autoimmune therapies.
|
|
Scooped by
mhryu@live.com
Today, 2:48 PM
|
Microbiologically induced concrete corrosion (MICC) poses a severe challenge to the long-term durability of infrastructure, particularly in sewer networks and marine environments, which is driven by microbial metabolic activities that attack cement hydrates (Ca(OH)2, C-S-H) mainly caused by biogenic sulfuric acid (from sulfur-oxidizing bacteria) or organic acids (from fungi), converting them into expansive gypsum and ettringite, and then cause cracking and spalling. This article reviews advances in mechanisms, key microorganisms, and protection strategies of MICC to enhance our understanding of MICC and provide a guideline for effective protection. The corrosion mechanisms differ by environment: sewers exhibit three-stage pH-driven succession, marine biofilms can either accelerate or inhibit corrosion, while fungi dominate in agricultural and historical settings. Core functional microorganisms involved in MICC include sulfur-oxidizing bacteria (SOB), sulfate-reducing bacteria (SRB), and acid-producing fungi (AF), following pH-dependent succession, while indicator microorganisms for protection efficacy include typical SOB, SRB, and AF that are involved in MICC, as well as general antimicrobial indicator strains (E. coli and Staphylococcus aureus) which are used only to assess broad antimicrobial activity and do not represent MICC-specific resistance. Multi-scale deterioration proceeds from microstructural decalcification and pore coarsening to macroscopic mass loss and compressive strength reduction. Protection strategies are categorized into: (i) corrosion-resistant materials (e.g., calcium aluminate cement and alkali-activated materials), (ii) antimicrobial additives (e.g., nano-ZnO and Cu2O), (iii) surface coatings (e.g., superhydrophobic coatings and electrodeposited Cu/Cu2O layers), and (iv) ecological regulation. However, significant gaps remain between laboratory efficacy and field performance, highlighting the need for long-term validation, multi-scale characterization, intelligent responsive materials, eco-compatible protection systems, and standardized microbial exposure systems.
|
|
Scooped by
mhryu@live.com
Today, 1:35 PM
|
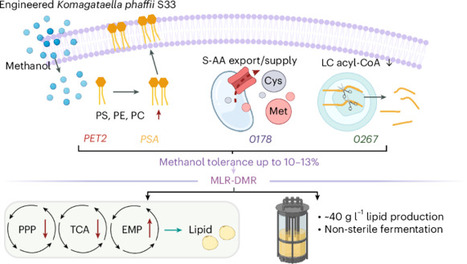
Conventional production of food lipids is environmentally damaging and may struggle to meet growing global demand, as many solid fats derive from animal sources and many plant oils depend on monocultures and tropical crops. Here we evaluate whether microbial production of food lipids from renewable carbon sources could provide a viable alternative using a conceptual modeling study that estimates process performance, costs, and resource availability. The analysis examines lipid-accumulating microbes grown on methane and methanol derived from organic waste, focusing on the production of phospholipids and triacylglycerols and estimating feedstock availability in Europe using published data and market prices. We estimate minimum selling prices of 14.2 United States dollars per kilogram for phospholipids and 10 United States dollars per kilogram for triacylglycerols, with costs mainly driven by carbon source, labor, electricity, and capital, suggesting potential economic and technical feasibility under favorable assumptions. Microbial phospholipids and triacylglycerols reach minimum selling prices of 14.2 and 10 US dollars per kilogram, according to a conceptual modeling study of methane- and methanol-based lipid production using European waste-derived feedstocks.
|
|
Scooped by
mhryu@live.com
Today, 10:47 AM
|
While engineering modular polyketide synthases (PKSs) using the recently updated module boundary has yielded libraries of triketide–pentaketides, this strategy has not yet been applied to the combinatorial biosynthesis of macrolactones or macrolide antibiotics. Here we developed a two-plasmid system for the construction and expression of PKSs and used it to obtain a refactored pikromycin PKS in E. coli that produces 49 mg of narbonolide per litre of culture. The replacement, insertion, deletion and mutagenesis of modules enabled access to hexaketide, heptaketide and octaketide derivatives. Supplying enzymes for desosaine biosynthesis and transfer enabled production of narbomycin, pikromycin, YC-17, methymycin and five derivatives thereof. By using native docking domains, knocking out pathways competing with desosamine biosynthesis and supplying the editing thioesterase PikAV, the titre of narbomycin was boosted 58-fold to 37 mg l−1. The second and third modules of the refactored pikromycin synthase were swapped with others to yield three new narbomycin analogues, investigate structure–activity relationships and demonstrate how libraries of macrolide antibiotics can be readily accessed. A biosynthetic platform is developed to rapidly construct large, engineered polyketide synthases using the updated module boundary. This system produces a library of macrolide antibiotics in Escherichia coli.
|
|
Scooped by
mhryu@live.com
June 28, 1:10 PM
|
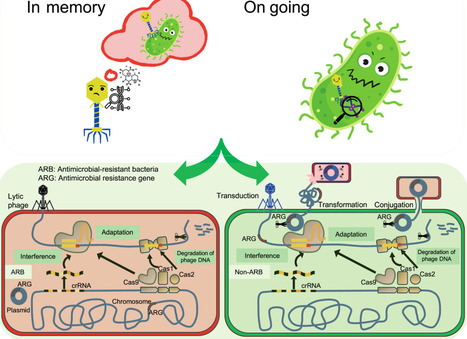
Phage–host interactions critically shape environmental antimicrobial resistance (AMR). Using swine manure anaerobic digestion and multi-omics (metagenomics, meta-transcriptomics, and Hi-C), we mapped the phage–bacteria arms race and its impact on AMR dynamics. We revealed that phage-mediated lysis overwhelmingly dominates transduction, while phages rarely carry antimicrobial resistance genes (ARGs), and phage-borne ARGs showed no expression, challenging the paradigm of phages as primary vectors of ARGs. Crucially, the intense on-going phage–host arms race drives the widespread presence and expression of antiviral defense systems (ADSs) in antimicrobial-resistant bacteria (ARB). These ADSs exhibit a vital ecological dual role: they protect ARBs from phage lysis promoting persistence while simultaneously suppressing horizontal gene transfer (HGT, e.g., conjugation), as validated by in vitro conjugation assays. Our findings elucidate this duality, offering a novel framework to harness phage lytic pressure and ADS-mediated HGT suppression for environmental AMR mitigation.
|
|
Scooped by
mhryu@live.com
June 28, 1:03 PM
|
Computational predictors of protein-binding sites within intrinsically disordered regions (IDRs) show highly inconsistent performance across high-quality benchmark datasets. To understand the origins of these discrepancies, we systematically compared predictors across three independent test sets: two CAID datasets updated with the latest DisProt annotations and a composite dataset (DBs) assembled from DIBS, FuzDB, IDEAL, and MFIB. Predictors trained predominantly on DisProt data achieved substantially higher AUCs on the CAID sets but performed poorly on the DBs. In contrast, predictors trained on older, low-quality PDB-based datasets showed balanced performance across all sets, with a slight preference for DBs. Predictors with mixed training exposure displayed intermediate behavior. Through controlled experiments using identical CNN architectures and feature analysis, we demonstrate that the dominant factor driving these performance differences is the intrinsic disorder propensity of the binding sites themselves. Binding residues in DisProt-based datasets exhibit markedly higher average disorder propensity scores than those in PDB-derived datasets. This previously unrecognized selection bias (literature studies preferentially characterizing more disordered binding sites, while PDB-derived annotations capture less disordered ones) effectively splits IDR-protein binding sites into two distinct categories. Predictors optimized on one category therefore generalize poorly to the other. Binding-site length and sequence conservation play only minor or negligible roles in explaining the observed inconsistencies. These findings highlight a critical limitation in current benchmarking practices and training strategies for IDR-binding site prediction, underscoring the need for more balanced and disorder-aware reference datasets. Finally, the diagnostic techniques introduced here could prove valuable beyond the specific application examined in this study.
|
|
Scooped by
mhryu@live.com
June 28, 12:48 PM
|
With the rise of antimicrobial resistance, urinary tract infections (UTIs) have become increasingly more difficult to treat, prompting renewed interest in bacteriophage (phage) therapy as an alternative or adjunct to antibiotics. UTIs are an attractive target for phage therapy because they generate a high density of actively replicating bacteria that supports phage propagation, and because the urinary tract is readily accessible for administration and monitoring. Yet studies of phage therapy for UTIs report mixed outcomes, including failures to meet clinical and microbiological endpoints. Here we follow the population dynamics of a clinical Escherichia coli UTI strain and two phages, HP3 and ES19, to which the strain appears susceptible by standard testing. Despite this apparent susceptibilty, both phages fail to suppress the strain, with resistance emerging almost immediately. Using the measured mutation rate, our mathematical model shows that traditional resistance cannot account for these dynamics. We instead demonstrate, including by a phage-specific population analysis profile assay we developed, that heteroresistance drives this rapid failure, offering a plausible explanation for treatment failures in UTI phage therapy
|
|
|
Scooped by
mhryu@live.com
Today, 5:21 PM
|
Microgravity is a condition that may affect gastrointestinal function and metabolism during spaceflight. Despite attempts to keep normal dietary habits at the ISS, the food selection and meal timing may also be perturbed during the confinement. To understand the impact of spaceflight on human metabolism, we characterized the plasma metabolome of astronauts (n = 52) before, during, and after missions on board the International Space Station (ISS) using liquid chromatography coupled with mass spectrometry. Here we show that spaceflight affected around 40 circulating compounds. In this longitudinal assessment, the metabolic changes were already observed shortly after launch and subsided within a few days after landing. The flight-induced changes in metabolites reflected increased protein fermentation by the gut microbiota, possibly reflecting prolonged intestinal transit time caused by microgravity. Minor diet-related changes related to intakes of caffeine, fish, and some fats were also observed but affected less than one third of all metabolites responding to spaceflight. We did not observe major sex-specific differences in metabolism. Increasing the consumption of slow-fermented carbohydrates at ISS to reduce protein fermentation might improve gastrointestinal health in astronauts in future human long-term spaceflights. Here, by metabolomics profiling of blood samples collected before, during and after spaceflight in 52 astronauts, the authors identify changes in gut microbial metabolites indicative of slower gut transit during microgravity.
|
|
Scooped by
mhryu@live.com
Today, 5:14 PM
|
The construction of minimal-genome microbes offers an ideal platform for understanding fundamental biological processes and synthetic biology, yet the research is hindered by incomplete lists of essential genes in microbes and by multiple rounds of genome trimming with a trial-and-error nature. To address this, we introduce CREAT (CRISPR-based genome trimming with a multi-homology-arm template)—a streamlined approach that integrates CRISPR-targeted genome cleavage and homology arm walking to classify essential from non-essential genomic subregions, thus providing the basis for predicting essential genes in a given organism. These essential genes were then assembled into synthetic gene cassettes for one-step replacement of the targeted non-deletable genomic regions for further genome trimming. Eight consecutive rounds of CREAT genome trimming achieved a 20.8% reduction in genome size in Saccharolobus islandicus. Furthermore, Cas9-based CREAT genome trimming was developed for Bacillus subtilis and E. coli, with efficiency greatly enhanced by the λ-Red recombinase in the latter. Together, this iterative application of CREAT provides a scalable and generally applicable strategy for rapidly constructing minimal genomes across diverse microorganisms.
|
|
Scooped by
mhryu@live.com
Today, 5:01 PM
|
Plant roots are broadly colonized by endophytic fungi with saprotrophic capabilities, but our understanding of whether they function in ways that are beneficial or detrimental to the host remains limited to model organisms. We hypothesized that endophytic fungi broadly affect plant access to soil nutrients, particularly organic forms that are typically not directly available to the plant. To address this, we paired 41 fungal endophytes with switchgrass (Panicum virgatum L.) and provided either inorganic or organic forms of nitrogen (N) and phosphorus (P). We evaluated how the fungi affected plant tissue N and P as well as plant growth. We also examined if these outcomes could be predicted from fungal phylogenetic relationships, in vitro traits of the fungi, or characteristics of the habitat from which fungi were isolated. There was substantial variation in both plant N (0.05-0.63%) and P (0.02-0.10%) acquisition that depended on the interaction of fungus and nutrient treatment. More fungi were beneficial for plant N than for P and shoot nutrients generally increased more than root nutrients from fungal associations. However, fungal effects on plant nutrients were not predicted by fungal traits, habitat traits, or fungal phylogenetic relationships. This unpredictability highlights a key challenge for incorporating endophytes into nutrient management strategies. Improving our ability to predict endophyte impacts on host nutrient acquisition will require identifying the mechanisms underlying observed beneficial effects and scaling up to realistic, diverse root microbial communities.
|
|
Scooped by
mhryu@live.com
Today, 4:53 PM
|
Microbial communities spontaneously colonize pristine environments, yet how species growth kinetics and individual cell variation shape community assembly remains poorly understood. Here, we use time-lapse microscopy imaging to track the division of individual founder cells in communities composed of up to seven soil bacterial isolates grown on nutrient surfaces. With cell lineage tracking, we quantify species-specific absolute biomass formation and growth kinetics from early growth through stationary phase. The reproductive success of individual founder cells depended on the timing of their first cell division, which determined their access to the primary substrate and their maximum growth rates. In mixed-species communities, founder cell success also depended on species-specific, substrate-dependent growth rates and yields. In addition, spatial factors – such as cells’ positioning, distances to non-kin neighbors, and identities of co-occurring species – further influenced outcomes. In spatially structured communities, interspecific interactions were globally governed by competition for primary substrates. We also observed cross-feeding of leaked metabolites, reflected in fluctuating paired interaction strengths and interaction signs. Species-pair interactions differed locally, with cells within distances of less than 15 µm exhibiting opposite interaction behaviors. Global pairwise interactions predicted from monoculture growth kinetics were observed in approximately half of the measured pairs, whereas measured paired interactions generally weakened in combinations of three or more species. Using a spatially explicit agent-based Monod growth model that includes interspecific interactions, we accurately predicted the compositions of seven-member communities. Overall, our results indicate that emergent, spatially mediated interspecific interactions between cells of different bacterial species primarily drive local and temporal changes in individual cell growth rates, which in turn determine final biomass formation. Because most natural microbial habitats are spatially structured, stochastic founder-cell positioning and fitness differences are key determinants of locally formed interaction patterns and species coexistence.
|
|
Scooped by
mhryu@live.com
Today, 4:18 PM
|
Genome-scale metabolic models (GSMMs) are important aids towards system-level understanding of the metabolic physiology of the gut microbes and for rational microbiome engineering. While large-scale repositories of GSMMs for gut-associated bacteria are available, strain-level variability and the continuous discovery of novel taxa through metagenomics and culturomics underscore the need for scalable, ab initio reconstruction tools. Here, we present CarveMe-GutMicrobes, a client-side framework for rapid reconstruction of metabolic models directly from (meta)genomic input. Building upon the original CarveMe framework, CarveMe-GutMicrobes incorporates an expanded, gut-microbe-centric biochemical database that includes reactions, metabolites, and gene-protein-reaction (GPR) associations curated specifically for Bacteria and Archaea inhabiting the human gut. The tool supports taxonomic restriction of the reference database to improve context-specific accuracy. To test the CarveMe-GutMicrobes and to address the paucity of experimental data for non-model gut taxa, we generated new experimental datasets on metabolite secretion profiles and gene essentiality. CarveMe-GutMicrobes models demonstrated high predictive performance performance against these as well as previously available datasets. By integrating curated resources, extending reaction coverage, and offering new empirical datasets, CarveMe-GutMicrobes provides a scalable platform for high-resolution metabolic reconstruction towards broader adoption of GSMMs in gut microbiome research.
|
|
Scooped by
mhryu@live.com
Today, 4:12 PM
|
Bacteria carry a large repertoire of antiviral defence systems, our knowledge of which is expanding rapidly. Several bioinformatics tools now exist to identify them. Though powerful, these tools can differ in the models they use and the nomenclature they return, thus a single tool could both miss an annotation and disagree with its peers. Here we describe Ptolemaea, a pipeline for harmonizing phage-defence annotations across multiple tools by reconciling PADLOC, DefenseFinder, and a bidirectional BLAST. Over a common predicted set of proteins, Ptolemaea provides a consensus annotation list per genome. The pipeline is not intended to outperform or replace its component tools; its purpose is to maximize the number of defence systems recovered from a genome and to make disagreements between tools explicit and resolvable. We demonstrate the pipeline on 700 complete genomes spanning the ESKAPE pathogens and E. coli, recovering 32,509 defence annotations, of which 50.6% were supported by more than one annotation source.
|
|
Scooped by
mhryu@live.com
Today, 3:02 PM
|
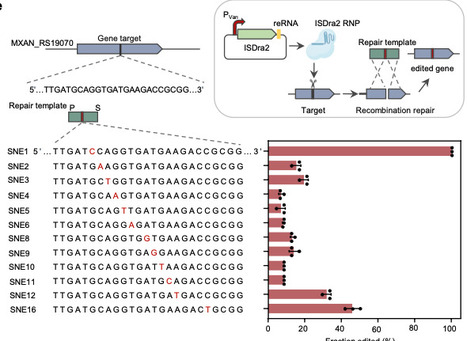
Myxobacteria are renowned producers of structurally diverse bioactive natural products. Motivated by a comprehensive inventory of myxobacterial biosynthetic gene clusters (BGCs), we develop efficient genetic toolkits and construct a robust heterologous expression chassis. We identify a pair of Myxococcus-derived recombinases, designated MxRecET, that enables efficient genome editing in the model strain Myxococcus xanthus DK1622. The synergistic combination of MxRecET recombineering with the transposon-associated nuclease TnpB (ISDra2) enables versatile genetic manipulations, including seamless deletion of large DNA fragments (up to 200 kb), tandem editing of dual loci, and flexible single-nucleotide substitutions within a streamlined two-week workflow. This integrated technology, termed MxDIRECT, facilitates the rational engineering of DK1622 into a plug-and-play chassis designated MxPKS, featuring enhanced growth properties, elimination of competing pathways, an improved precursor supply, and increased robustness. The effectiveness of MxPKS is demonstrated through heterologous expression of four polyketide synthase BGCs, leading to the discovery of multiple unknown polyketides. This work establishes a foundation for accelerated bioprospecting of myxobacteria. Myxobacteria are renowned producers of structurally diverse bioactive natural products but are difficult to engineer. Here the authors create a robust heterologous expression chassis and develop MxDIRECT, a recombineering strategy for scarless marker-free expression of biosynthetic gene clusters.
|
|
Scooped by
mhryu@live.com
Today, 2:29 PM
|
Enhanced production of isoprenoid compounds, including carotenoids, is needed to meet the growing demand in the food, cosmetic, pharmaceutical, and biotechnology sectors. Haloferax volcanii represents a promising microbial platform for sustainable isoprenoid production, as this halophilic archaeon is well suited for metabolic engineering, thrives under harsh conditions (UV irradiation, high temperatures, and metal-induced stress) compatible with bioprocessing, and naturally synthesizes carotenoids including the high-value C50 bacterioruberin. In this study, we optimized carotenoid yield in H. volcanii using a chemically defined medium supplemented with cost-effective, industrially favorable feedstocks of crude glycerin and urea as the sole carbon and nitrogen sources, respectively. Following optimization by reuse of medium and supplementation with additional glycerin, carotenoid production was evaluated. Transcript abundance of the carotenoid 3,4-desaturase gene (crtD, HVO_2528) was measured, and β-galactosidase (bgaH) reporter activity was used to assess crtD promoter activity. H. volcanii was found to display comparable growth rates on crude glycerin to laboratory grade glycerol. Following optimization by reuse of medium and supplementation with additional glycerin, an 8-fold increase in carotenoid yield was observed when urea (84.1 ± 8.4 mg⋅L−1) served as the nitrogen source compared to cultures only grown with crude glycerin and NH4Cl (10.0 ± 2.4 mg⋅L−1) when normalized. The higher carotenoid yield on urea vs. NH4Cl was found to be correlated with a 3- to 4-fold increase in transcript abundance of the carotenoid 3,4-desaturase gene (crtD, HVO_2528) that was regulated at the level of transcription. The crtD promoter was therefore identified as a strong candidate based on β-galactosidase (bgaH) reporter activity for use in metabolic engineering. Urea offers higher nitrogen content, reduced acidification potential, and greater scalability than NH4Cl for bioprocessing applications. Together, our findings support the development of a sustainable, circular approach for repurposing industrial glycerin waste streams to support carotenoid production using urea as a nitrogen source and H. volcanii as a microbial biocatalyst for renewable biomanufacturing.
|
|
Scooped by
mhryu@live.com
Today, 1:29 PM
|
Serine integrases can precisely integrate large DNA constructs into desired chromosomal sites but only if their natural target site is first installed into the recipient genome. Here, to retarget serine integrases to a desired genomic site, we develop a modular integrase (MINT) system for genome editing. Through a combination of structural modeling, single-round directed evolution and screening in human cells, we retargeted the specificity of the serine integrase Bxb1. We demonstrate the therapeutic potential of the MINT system by retargeting Bxb1 to the human AAVS1 and TRAC loci, where wild-type Bxb1 has no detectable activity. By combining MINT constructs with both known activity-increasing Bxb1 mutants and zinc-finger DNA-binding domains, we achieve efficiencies of 29% at the AAVS1 locus and 35% at the TRAC locus in K562 cells. To further demonstrate clinical potential, we achieved 29% GFP integration efficiencies at the TRAC locus in human T cells. Serine integrases are retargeted to streamline genomic integrations of DNA.
|
|
Scooped by
mhryu@live.com
Today, 10:44 AM
|
Ecosystems are shaped by communities of microorganisms whose niches and impacts depend on functional profiles influenced by gene gains and losses. Culture-based experiments demonstrate that mobile genetic elements (MGEs) can mediate gene flux, but quantitative understanding of these dynamics in natural systems remains limited. Here we develop and apply a systematic, meta-omic framework to investigate MGEs in a complex natural system using an 8-year soil time series collected at Stordalen Mire, in Sweden’s thawing permafrost margin. In this climate-critical peatland, we identify ~2.1 million MGE recombinases across 89 microbial phyla and assess ecological distributions, affected functions, past mobility and current activity. This revealed an active mobilome that shapes natural genetic diversity via differential impacts on major phyla and affects a wide range of functions, including metabolic genes involved in carbon flux and nutrient cycling. These findings and this analytic framework suggest avenues towards a better understanding of MGE diversity, activity, mobility and impacts across ecosystems. An 8-year soil meta-omic time series shows how mobile genetic elements shape permafrost microbial diversity and impact a range of functions, including carbon and nutrient cycling.
|
|
Scooped by
mhryu@live.com
June 28, 1:06 PM
|
Designing binders against novel protein targets remains a central challenge in computational drug discovery. Here we introduce BoltzProt-1, a pipeline for generating protein binders, including nanobodies, with improved hit rates and favorable developability properties. At its core lie a refined iteration of BoltzGen's generative model and a novel protein-protein interaction prediction model, BoltzPPI. Employing BoltzPPI instead of BoltzGen's standard structure-prediction confidence metrics to rank nanobody (VHH) designs increases the confirmed-binder hit rate from 3.3% to 8.0% across 10 novel targets. Assessed on 10 additional targets used in prior literature, the BoltzProt-1 pipeline obtains nanobody screening hits for 7 of 10 targets, surpassing the 6 of 10 previously reported by Chai-2. Finally, evaluating the developability of BoltzProt-1-designed nanobodies in terms of stability, aggregation, purity, polyspecificity and hydrophobicity reveals that 58% of its confirmed binders pass every criterion, exceeding both BoltzGen (40%) and clinical-stage VHH controls (21%).
|
|
Scooped by
mhryu@live.com
June 28, 12:56 PM
|
The 5′ untranslated region (5′ UTR) is a key regulatory element that governs mRNA translation and protein output. However, existing computational methods typically address isolated tasks such as functional prediction or sequence optimization, limiting their ability to support rational design across the full 5′ UTR engineering workflow. Here, we present UTRGen, a unified modeling framework for 5′ UTRs that integrates sequence generation, multi-property prediction, and constrained function-guided design. UTRGen is pre-trained autoregressively on large-scale 5′ UTR datasets from multiple species and subsequently adapted to diverse downstream regulatory tasks. Across systematic evaluations, UTRGen generates novel and diverse 5′ UTRs while preserving sequence, structural, and functional characteristics of natural UTRs. After task-specific fine-tuning, UTRGen achieves state-of-the-art performance across 14 benchmark datasets, improving translation efficiency prediction by up to 11.1%, expression level prediction by up to 13.2%, and mean ribosome load prediction by up to 3.0% relative to the strongest baselines. It also achieved the best overall performance for internal ribosome entry site identification. To enable controllable design, we formulate function-guided 5′ UTR design as a GRPO-based refinement process over a pre-trained autoregressive sequence prior, using composite rewards to encode functional objectives and biological constraints while regularizing toward the natural 5′ UTR distribution. The resulting sequences show consistently improved predicted translation efficiency and expression levels across cellular contexts, and reveal interpretable sequence features associated with high activity, including reduced C content, fewer upstream AUGs, and depletion of inhibitory motifs. Together, our results establish a unified modeling strategy for 5′ UTR design and lay a foundation for programmable control of translation.
|