 Your new post is loading...

|
Scooped by
mhryu@live.com
May 6, 1:09 PM
|
Can synthetic biology provide food security in a changing climate?
|
|
Scooped by
mhryu@live.com
May 6, 12:22 PM
|
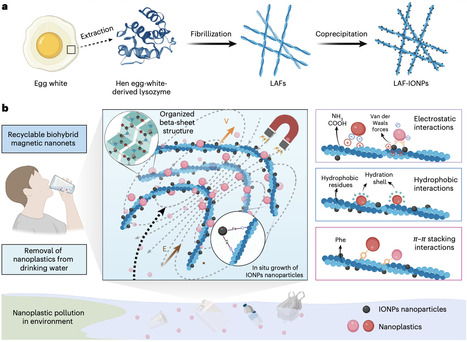
Nanoplastics (NPs), as persistent and unregulated aquatic contaminants, evade conventional removal technologies, posing severe health risks and urgently demanding innovative, effective solutions. Here we introduce recyclable magnetic biohybrid nanonets (LAF-IONPs), engineered through in situ growth of iron oxide nanoparticles (IONPs) on lysozyme amyloid fibrils (LAFs). Stabilized by synergistic interfacial interactions, LAF-IONPs efficiently capture NPs over a wide range of sizes (30–1,000 nm) and chemical compositions across environmentally relevant concentrations and conditions (pH 7–9, high salinity and co-existing pollutants). The superior removal efficiency arises from magnetic active motion and abundant fibril binding sites. Remarkably, LAF-IONPs achieved 98.0–99.9% NPs removal from various real water samples and maintained >95% efficiency over 100 recycling cycles when using a custom alternating magnetic field system. Critically, LAF-IONPs treatment reduced in vivo NPs bioaccumulation by 91.5%. This work establishes a blueprint for designing recyclable biohybrid adsorbents for active, efficient and sustainable removal of NPs and potentially other emerging contaminants. Nanoplastics are persistent water contaminants that evade conventional removal methods. Magnetic biohybrid nanonets are found to capture up to 99.9% of them, remain highly recyclable and greatly reduce in vivo accumulation.
|
|
Scooped by
mhryu@live.com
May 6, 10:19 AM
|
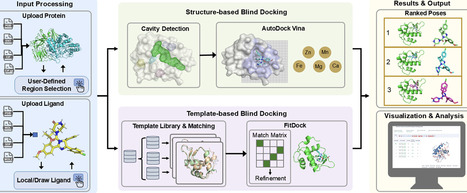
Elucidating protein–ligand interactions is pivotal for understanding biological mechanisms and accelerating drug discovery. Blind docking, which identifies binding sites without prior knowledge, has become an indispensable computational strategy for analyzing the surge of protein structures generated by Cryo-EM and AI-based prediction tools like AlphaFold3. Our previous server, CB-Dock2, has been widely adopted by the global research community, averaging over 1000 daily submissions since July 2022 due to its accuracy and user-friendliness. Building on this foundation and incorporating extensive user feedback, we present CB-Dock3, a substantially enhanced platform. Key upgrades include a refined docking engine, an expanded template library, and support for diverse file formats. Benchmark evaluations on CASF-2016 demonstrate that CB-Dock3 achieves a success rate of 67.4% (RMSD ≤ 2.0 Å), representing a 10.6 percentage-point absolute improvement over its predecessor and outperforming other popular blind docking tools. Additionally, CB-Dock3 introduces critical new features driven by community needs: support for user-defined docking regions to handle large complexes, and a metal-aware protocol that explicitly retains essential metal ions and cofactors during simulation.
|
|
Scooped by
mhryu@live.com
May 6, 10:09 AM
|
A wide range of microbially produced specialized metabolites (SMs) with bioactivity affecting micro- and macro-organisms has been described. These bioactive compounds are synthesized by enzyme complexes encoded in biosynthetic gene clusters (BGCs). Historically, the first role of SMs was revealed through antimicrobial activity-based assays. However, diverse bioactivities have since been deciphered that are not associated with inhibition of other organisms. To advance discoveries in chemical ecology, synthetic microbial communities (SynCom), simplified, experimentally tractable systems that recapitulate specific features of natural microbiomes, are increasingly employed. SynComs enable the systematic investigation of SM-mediated induction of BGC expression, the characterization of SM bioconversions, and the elucidation of the mechanism by which SMs influence microbial establishment and persistence within communities. Due to their reduced complexity, SynComs allow the controlled determination of microbial community composition and functional dynamics, as well as the characterization of associated chemical diversity. This review highlights representative publications describing how SynComs are employed to elucidate the roles of SMs in complex microbial interactions and emphasizes the emerging functions observed in SynCom-based chemical ecology studies.
|
|
Scooped by
mhryu@live.com
May 6, 9:39 AM
|
Knowledge of the regulatory mechanism of riboswitches is vital for understanding how microorganisms cope with changes in both intracellular and extracellular environments and for developing and applying RNA biosensors. To date, two types of glutamine-based riboswitches, which are exclusively distributed in cyanobacteria, have been identified. Here, we found an RNA regulatory element in the 5′UTR of the nucleoside permease gene (nupC) in Bacillus thuringensis BMB171; it was identified as a novel glutamine riboswitch and named LRN (leader RNA of nupC). Unlike the two previously known types of glutamine riboswitches found in cyanobacteria, LRN is a single-domain RNA element representing a novel type III glutamine riboswitch. Binding glutamine leads to rearrangements the LRN RNA structure, which inhibits downstream gene expression at the transcriptional level. Biocomputational searches revealed that LRN is frequently found in the Bacillus cereus group and is located mainly upstream of the coding region of the nupC homologues. Thus, this RNA-based sensing mechanism establishes a regulatory feedback loop that couples intracellular glutamine levels to nucleoside transport, which is shared by the B. cereus group.
|
|
Scooped by
mhryu@live.com
May 6, 1:42 AM
|
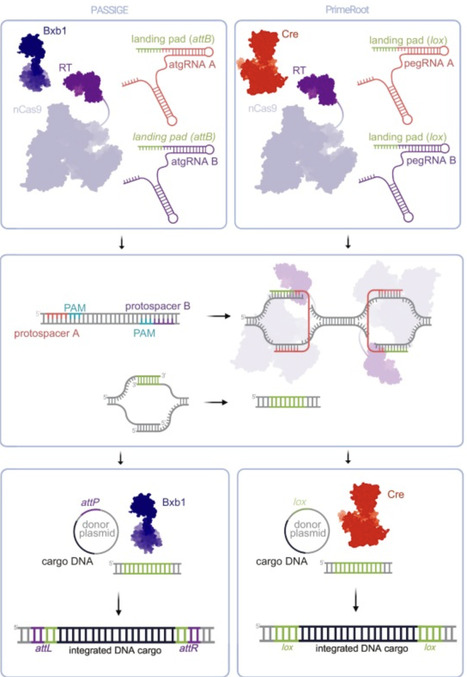
While the advent of CRISPR-based technologies has democratized the genesis of precise mutations, there is a need for more sophisticated tools to enable large-scale DNA manipulations, advancing genome editing across medicine, biotechnology, and agriculture. The success of Cas9 and Cas12 has hinged on the generation of precise DNA nicks and double-stranded breaks (DSBs), enabling local sequence mutagenesis, albeit of a limited size range. Emerging effectors combining Cas with other enzymatic functions, such as CRISPR-associated transposons and site-specific recombinases, enable larger integrations. Sophisticated combinations such as programmable addition via site-specific targeting element (PASTE), prime-editing-assisted site-specific integrase gene editing (PASSIGE), and prime-editing-mediated recombination of opportune target (PrimeRoot) expand payload options and DSB-free editing modalities, with translational potential for next-generation crop breeding in sustainable agriculture and the development of gene and cell therapies in personalized medicine.
|
|
Scooped by
mhryu@live.com
May 6, 1:14 AM
|
L-xylulose is a versatile L-form rare sugar with exceptional properties, including low caloric content, α-glucosidase inhibition, and antiviral effects, making it highly valuable for functional food and pharmaceutical applications. However, natural L-xylulose is extremely scarce, and its chemical synthesis is plagued by harsh conditions and isomer contamination, thus making biosynthesis—with its industrial potential and environmental friendliness—a promising alternative. This review summarizes the latest biosynthesis progress, analyzes six enzymatic pathways for L-xylulose and identifies two competitive routes, using xylitol and L-arabinitol as starting substrates. It further elaborates on core bottlenecks in chassis cell construction and industrial fermentation of L-xylulose, emphasizing that breakthroughs rely on three strategies: AI-driven high-yield strain screening, catalytic optimization of key enzymes, and efficient extracellular transport/secretion of the target product. Finally, the integration of synthetic biology with intelligent biomanufacturing is proposed as a crucial direction for efficient L-xylulose biosynthesis, facilitating its broader application across the food, pharmaceutical, and chemical industries.
|
|
Scooped by
mhryu@live.com
May 6, 12:24 AM
|
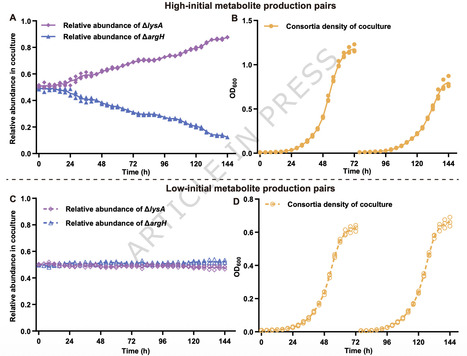
Syntrophic interactions based on reciprocal metabolite exchange are widespread in microbial communities, yet the factors determining their stability remain unclear. Using synthetic E. coli consortia composed of lysine and arginine auxotrophs, we show that lower initial metabolite production promotes, rather than limits, syntrophic stability. During serial propagation, replicate cocultures diverged sharply: a minority maintained sustained growth, whereas most became extinct. This divergence was associated with phenotypic differences in metabolite production among founding isolates. Consortia founded by low-producing strains recovered reliably after dilution and were more resistant to invasion by non-producing mutants. By contrast, high-producing founder generated diminishing returns for consortium growth, and increased extracellular metabolite availability that favored exploitation by non-producer. Although we detected no consistent coding-region variations between high- and low-producing isolates, expression differences suggest that outside coding regions may influence these production traits. These results identify constrained initial metabolite production as a key determinant of syntrophic stability. Low initial metabolite production stabilizes syntrophic E. coli consortia by constraining metabolite availability, promoting localized exchange, and resisting invasion by non-producers
|
|
Scooped by
mhryu@live.com
May 5, 11:46 PM
|
Gene order is a powerful design principle for protein nanomachines. In nature, gene organization ensures the precise assembly of functional protein nanostructures. We demonstrate how genetic repositioning of the key structural gene pduN, within the operon encoding a self-assembling protein nanocompartment, sculpts the morphology and function of bacterial microcompartments (BMCs). Relocating pduN to new operonic positions dramatically altered the size, shape, and catalytic output of BMCs, despite identical protein sequences. These shifts reveal how gene order may control nanoscale assembly and compartmentalised function. Our findings establish operon architecture as a programmable genetic framework for nanostructure morphogenesis and provide a synthetic biology strategy to engineer self-assembling nanodevices with customised geometries and activities.
|
|
Scooped by
mhryu@live.com
May 5, 11:30 PM
|
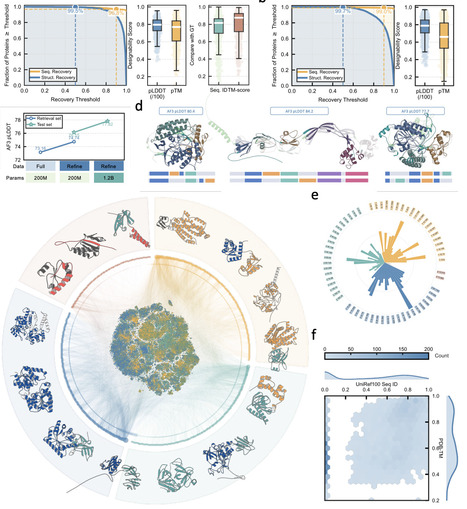
Multidomain proteins arise through the reuse and recombination of structural domains, yet natural architectures represent a sparse, structured sample of the possible domain-combination space. Here, we introduce DOMINO, a two-stage framework that learns domain co-occurrence from TED-annotated multidomain proteins and uses the learned patterns to generate new multidomain sequences. DOMIN, a contrastive retrieval model, embeds domains into a latent compatibility space and retrieves candidate partners for a query domain from a TED-derived domain pool, including pairings not observed in the TED-derived co-occurrence set. DOMO, a conditional autoregressive sequence model, converts each retrieved domain pair into a full-length protein sequence by jointly generating the specified domain regions and the non-domain sequence context between and around them. DOMIN recovers hierarchical patterns of natural domain co-occurrence and expands the observed CATH homologous-superfamily co-occurrence network with candidate novel pairings. DOMO realizes both held-out natural pairs and DOMIN-retrieved pairs as proteins with high domain recovery and high AlphaFold-predicted structural confidence. Applied at scale, DOMINO generated 5 million retrieval-derived multidomain proteins, with sampled designs showing recovery of the specified domains, diverse CATH annotations, and sequence novelty relative to UniRef100. Together, these results support domain co-occurrence as a predictive design prior and demonstrate a scalable strategy for exploring multidomain protein architectures through new combinations of existing structural modules.
|
|
Scooped by
mhryu@live.com
May 5, 11:10 PM
|
Medicinal plants link agriculture, ecosystem health, and human therapeutics, with bioactive compound profiles providing a direct and economically meaningful readout of microbiome function. Although microbial inoculation can enhance pharmacologically relevant metabolites under controlled conditions, these effects are context dependent and rarely reproducible in the field. This efficacy gap reflects three ecological constraints: introduced microbes are excluded by resident communities, environmental variation overrides laboratory-optimized functions, and inoculants fail to persist without mutualistic feedback. Addressing these barriers requires shifting from disposable inputs to microbiome stewardship: rewilding beneficial communities, designing climate-adapted consortia, and managing soil as living infrastructure. Whether such stewardship produces measurably different bioactive profiles and therapeutic outcomes under field conditions remains the empirical question on which its One Health rationale ultimately depends.
|
|
Scooped by
mhryu@live.com
May 5, 5:14 PM
|
Protein language models (pLMs) capture evolutionary sequence constraints but are limited in modeling underrepresented functional classes due to training data imbalance. Metalloproteins constitute a fundamental but sparsely represented class in sequence databases. We therefore assess whether structure-conditioned synthetic sequences can be used to specialize pLMs toward metal-binding functionality. We fine-tuned the generalist model ProtGPT2 on synthetic sequences generated by the inverse-folding model ProteinMPNN, constructing training sets with controlled variation in size and diversity. Fine-tuning increased recovery of canonical metal-binding motifs from 43% in the baseline model to 91% in the fine-tuned models. Generated sequences retained high predicted structural confidence and structural similarity to known folds, despite low sequence identity. Analysis of latent representations from ProtGPT2 indicated that fine-tuned models occupy distinct regions of embedding space relative to both the baseline model and structure-conditioned sequences, consistent with partial incorporation of structural constraints while preserving sequence diversity. A multi-step filtering pipeline applied to sequences lacking canonical motifs identified candidate metal-binding sites in four-helical bundle topologies not detected in a non-redundant subset of Protein Data Bank structures or in AlphaFold-predicted proteomes. https://doi.org/10.5281/zenodo.18672158 https://huggingface.co/gsgueglia
|
|
Scooped by
mhryu@live.com
May 5, 3:38 PM
|
Although several existing protein-protein interaction (PPI) databases provide yeast PPI data, none unify large-scale network topology information with detailed biophysical, proteostasis, and regulatory annotations in a single protein-centric framework. To address this gap, we developed the ANnotated Yeast Interactome (ANYI), an open, integrated resource that combines experimental yeast PPIs with sixteen feature annotation types, including protein abundance, half-life, disorder content, post-translational modifications, conformational stability, chaperone interactions, sequence, and structure. ANYI integrates 3,927 proteins with 155 annotation features, forming a unified matrix that enables systematic cross-layer analyses. Available via GitHub and Docker Hub with an interactive network browser for broad accessibility, ANYI provides both experienced and beginner computational scientists with tools to investigate the yeast interactome. For example, users can directly test whether highly connected hub proteins exhibit distinct stability, disorder, or proteostasis signatures relative to peripheral nodes.
|
|
|
Scooped by
mhryu@live.com
May 6, 12:37 PM
|
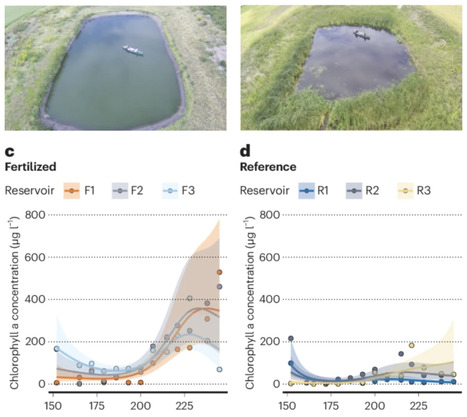
Urea, the world’s predominant nitrogen fertilizer, has supported human population growth for the past 60 years, yet its effects on freshwater ecosystems are largely unknown. Here urea additions at ecologically relevant rates tripled summer phytoplankton abundance in replicate agricultural reservoirs with significant responses by most eukaryotic algae, but not cyanobacteria or their toxins. Mass budgets reveal that fertilized reservoirs did not become limited by phosphorus due to its continuous release from sediments. Further, most added nitrogen did not accumulate in reservoirs but was lost to the atmosphere, probably as NH3. Sub-continental spatial analysis shows that study reservoirs are characteristic of shallow water bodies within Canada’s largest agricultural region, where >40% of surface waters are vulnerable to degradation by urea. Similar degrees of water quality loss by urea are expected in other global agricultural regions (for example, China, India, North America) where elevated urea use interacts with phosphorus-rich surface waters to induce extreme eutrophication. Urea, the most widely used nitrogen fertilizer, has unknown impacts on freshwater ecosystems. This study demonstrates that urea additions in Canadian prairie agricultural reservoirs triple phytoplankton abundance without increasing cyanobacterial toxins, revealing considerable nitrogen loss to the atmosphere and highlighting potential global water quality degradation in phosphorus-rich agricultural regions.
|
|
Scooped by
mhryu@live.com
May 6, 10:24 AM
|
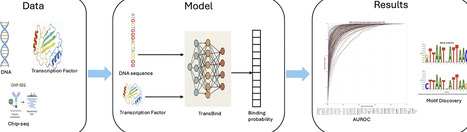
Transcription factors (TFs) regulate gene expression by binding to specific DNA sites on genome, making accurate TF binding site prediction critical for understanding gene regulation and downstream phenotypes. Almost all current deep learning methods use only DNA-related information to predict TF binding sites, ignoring the fact that different TF protein sequences and structures recognize distinct DNA patterns. Not leveraging TF information not only limits prediction accuracy but also makes the methods not generalizable to predicting binding sites of new TFs that do not exist in the training data. Here, we present TransBind, a protein-aware deep learning architecture that integrates DNA sequence information with protein embeddings containing both sequence and structural information derived from a protein language model pretrained on DNA-binding proteins, to improve TF binding site prediction. Through the cross-attention, a TF embedding selectively attends to genomic regions according to its unique binding properties. Evaluated on the data of 690 ChIP-seq experiments spanning 161 TFs across 91 human cell types, TransBind achieves an AUROC of 0.9508 and AUPR of 0.3741—representing a 11.8% relative AUPR improvement over state-of-the-art methods including TBiNet, EPBDXDNABERT-2, DanQ, and DeepSEA. The model outperformed existing methods in 98% of TF–cell type combinations. It also recovered 160 known TF binding motifs in the JASPAR database, providing the biological interpretability of the model. Moreover, the approach enables label-zero-shot prediction for unseen TFs, demonstrating its potential of generalizing to new, poorly characterized TFs. The source code of TransBind is available at https://github.com/jianlin-cheng/TransBind. The version used in this work is archived at https://doi.org/10.5281/zenodo.19462292.
|
|
Scooped by
mhryu@live.com
May 6, 10:17 AM
|
Optogenetic tools—light-responsive proteins that enable to regulate specific cellular activities, study biological processes, and develop new therapies—are attractive approaches for achieving endogenous gene regulation under minimally invasive conditions. Our first step in constructing an optogenetic system to regulate endogenous Drosophila gene expression was to identify inhibitory anti-CRISPR (Acr) proteins that block CRISPRa-mediated activation. Next, we inserted optogenetic protein LOV2 into these Acrs, tested for their ability to optogenetically modulate endogenous gene upregulation through the CRISPRa-based flySAM system in Drosophila, and found that the photoswitchability of these prototypes was weak. We therefore engineered an optimized Acr–LOV2 fusion module by refining length of intrinsically disordered and ordered regions (IDR and IOR) of Acrs. This optimization yielded a variant with significantly greater sensitivity to blue-light-induced endogenous gene upregulation than the prototypes, leading to new in vivo discoveries. In addition, this work provides insights for in vivo functional characterization of the IDR and the IOR of these small-sized proteins. Together, these findings establish a robust optogenetic toolbox for precise, light-controlled endogenous gene regulation in Drosophila.
|
|
Scooped by
mhryu@live.com
May 6, 10:05 AM
|
Bifidobacterium longum is a prevalent human gut symbiont whose carbohydrate metabolism is well characterized, whereas the quantitative contribution of amino acids to growth remains unclear. Here, we combined genome-based pathway analysis, growth phenotyping in chemically defined media, and iterative machine-learning-guided medium design to quantify amino acid preferences in B. longum subsp. longum JCM 1217T. Genome analysis predicted cysteine as the sole auxotrophy, and experiments confirmed that cysteine alone supported growth but did not restore the high maximum cell density and short lag time achieved with a complete amino acid mixture. Regression models and genetic algorithms identified amino acid formulations in the selected optimized compositions that reduced total amino acid input by 66.5% under glucose and 77.2% under lactose while maintaining growth comparable to complete medium. SHAP analysis highlighted tyrosine as the main determinant of maximum cell density, whereas glutamate, leucine, and valine consistently shortened lag time. These results show that amino acid requirements in B. longum extend beyond binary auxotrophy and provide a machine-learning framework for designing streamlined defined media.
|
|
Scooped by
mhryu@live.com
May 6, 1:45 AM
|
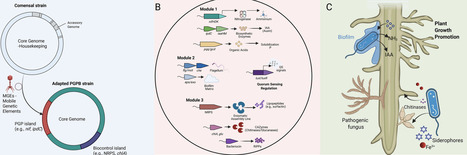
The demand for sustainable agriculture has shifted bioprospecting towards microbial bioinputs as alternatives to chemical fertilisers and pesticides. Whole-genome sequencing accelerates the discovery of plant-growth-promoting bacteria (PGPB) by enabling the identification of functional genes and the prediction of traits such as nutrient solubilisation, phytohormone production and biocontrol. Traditionally a secondary tool for strain characterisation, genomics has evolved into a ‘genome-first’ strategy that effectively collapses the phenotypic bottleneck in prospective bioprospecting and the rational design of synthetic microbial communities (SynComs). In this review, we argue for a transition from empirical phenotypic screening towards a genomics-guided paradigm for the selection of next-generation bioinputs. This work demonstrates how actionable insights can be gained through the integration of high-resolution genome mining into discovery pipelines. We explore the application of reverse ecology to infer ecological roles from genomic content and emphasise the critical role of pangenomics in identifying traits linked to host colonization and niche adaptation. Furthermore, we advocate for biosafety screening as a non-negotiable prerequisite for bioinoculant development to ensure ecological and clinical safety. Finally, this work proposes that genome-scale metabolic networks are essential to enable the transition from single-strain inoculants to the assembly of stable SynComs. This framework establishes a comprehensive, data-driven approach to predictable interventions in the agricultural bioeconomy.
|
|
Scooped by
mhryu@live.com
May 6, 1:20 AM
|
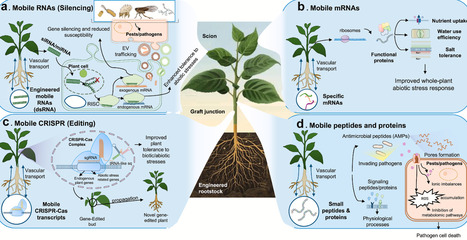
Grafting is a routine method widely used for the vegetative propagation of fruit trees and several vegetable crops. The selection of a compatible rootstock is essential, as it strongly influences scion growth, development, and resilience to biotic and abiotic stresses. It has been proven that messenger RNAs (mRNAs), small RNAs (sRNAs), proteins, and peptides move through the phloem and cross the graft interface, enabling direct communication between rootstock and scion. In particular, sRNAs and regulatory peptides have emerged as key mediators of stress resistance by modulating gene expression and activating systemic defence responses. In this review, we summarize genetic engineering approaches used to express target molecules, including CRISPR–Cas9-associated sequences, specifically in the rootstock, and we examine their effects following translocation to the scion. Engineered rootstocks have been shown to act as sources of mobile molecules that are transported to the scion, enhancing its tolerance to pathogens, pests, and environmental stresses without modifying the scion genome. Understanding the transport mechanisms and functional roles of these molecules represents a promising avenue for developing crops with reduced susceptibility to a wide range of biotic and abiotic stresses through advanced grafting technologies. Furthermore, this approach expands the potential of grafting as a biotechnological tool and represents a promising frontier also from a regulatory perspective.
|
|
Scooped by
mhryu@live.com
May 6, 1:04 AM
|
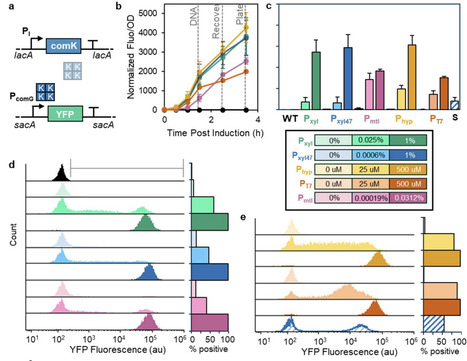
Bacillus subtilis is an important chassis for biotechnology, but its use in multiplex genome engineering is limited by low natural transformation efficiency. Here, we compared inducible promoter systems for synthetic activation of the competence regulator ComK and evaluated their effects on the comG operon competence reporter and transformation efficiency. Xylose- and mannitol-inducible systems outperformed IPTG-based constructs and shifted 96-99% of cells into a reporter-positive competent state. However, reporter activation alone did not predict transformation potential. Optimization of culture density and induction timing increased transformant yield 45-fold relative to the initial protocol and 2800-fold relative to the conventional Spizizen method. Disruption of native competence regulatory genes did not improve performance and often reduced transformation output, highlighting the importance of endogenous regulatory circuitry. Using the optimized strain and protocol, we achieved co-transformation frequencies of 11-18% and constructed multiplex spore-display libraries containing fluorescent protein fusions integrated at multiple loci. Screening identified strong dual-display combinations and showed that cargo loading depends on anchor protein, integration locus, and genetic background. SscA fusions supported the highest display capacity and promoted synergistic co-display. Together, these results show improvements in natural transformation-based genome engineering in B. subtilis and provide insight into the construction of multifunctional engineered spores.
|
|
Scooped by
mhryu@live.com
May 6, 12:11 AM
|
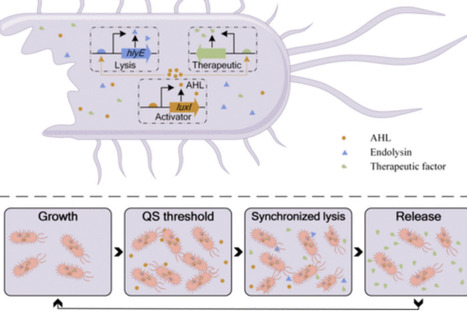
Synthetic biology has driven growing interest in engineered bacteria for cancer therapy. Salmonella Typhimurium stands out as one of the most promising live biotherapeutic products (LBPs) because of its innate tumor-targeting and colonizing abilities. However, the pathogenicity and inconsistent performance of wild-type S. Typhimurium have hindered further clinical translation. Recent advances in programmable and modular genetic redesign now enable precise reprogramming of bacterial functions, facilitating the creation of customized LBPs with enhanced safety and efficacy. In this review, we propose a systematic engineering framework with adaptive optimization and therapeutic enhancement for transforming S. Typhimurium into an effective anticancer LBP. This integrated strategy encompasses multiple facets, including strain attenuation, targeted enhancement, colonization optimization, therapeutic production, lysis-controlled release, and auxiliary modules. This review summarizes the key methodologies for engineering S. Typhimurium, highlighting its evolution from a pathogen to an antitumor vehicle. Furthermore, we summarize the multimodule collaborative engineering design and applications and call for more combinatorial strategies to enhance therapeutic efficacy. We also discuss the challenges and bottlenecks of engineered S. Typhimurium from the perspectives of genetic stability and clinical translation. Finally, we highlight how these synthetic biology advances are refining the mechanistic understanding of bacteria-mediated tumor therapy and paving the way toward safer, more effective, and clinically controllable anticancer strategies.
|
|
Scooped by
mhryu@live.com
May 5, 11:35 PM
|
The universe of possible protein sequences is astronomically large, yet our understanding of the sequence-structure relationship is confined to the infinitesimal fraction used currently by life. Determining whether "foldable" architectures are rare singularities or accessible solutions is critical for understanding protein evolution and designing novel proteins. Here, we map the structural landscape of random sequence space by screening one million synthetic proteins using a high-throughput in vivo FRET biosensor. We reveal that this space is structurally heterogeneous, populated not only by disordered chains and stress-inducing aggregates but also by "benign" compact structures that resemble globular proteins and evade cellular chaperone responses. By training machine learning models on these phenotypes, we show that structural potential is learnable and generalizes to natural proteomes. These findings demonstrate that biology-like folds are accessible from random sequences with surprising frequency, providing data required to expand generative protein design beyond evolutionary priors.
|
|
Scooped by
mhryu@live.com
May 5, 11:18 PM
|
Despite considerable powers, the application of CRISPR activation (CRISPRa) screens in primary human cells remains a formidable challenge. Here, we develop dCas12f1-SAM, a compact SAM-based transcriptional activation platform, that outperforms existing systems in both immortalized cell lines and primary human T cells and hematopoietic stem/progenitor cells (HSPCs). Using dCas12f1-SAM, we perform a pooled CRISPRa screen targeting 1559 human transcription factors (TFs) in primary human T cells and identify multiple positive regulators of IL-2 expression. We further implement a single-cell CRISPRa screen via our miCROP-seq construct, resolving how these genetic perturbations reshape T cell activation dynamics and drive functionally distinct cellular states. Among the top-ranking genes, we spotlight KLF12 and LHX5, whose overexpression significantly improves antigen-specific responses of chimeric antigen receptor T (CAR-T) cells. Collectively, these findings establish dCas12f1-SAM as a robust transcriptional activation tool, highlighting its potential to advance applications in cellular engineering and immunotherapy. CRISPRa screening in primary human cells has remained a formidable technical challenge. Here, the authors developed dCas12f1-SAM and successfully implemented a gain-of-function screen in primary human T cells, identifying key regulators of T cell activation.
|
|
Scooped by
mhryu@live.com
May 5, 5:18 PM
|
Biological information can be encoded in signaling dynamics, which have been implicated in many physiological processes; yet the diversity of dynamic expression profiles driven by a single gene remains unclear. To explore this, we screen 80 chromatin-associated proteins (CAPs) for their potential to drive diverse dynamic gene expression profiles from the same genome-integrated reporter in yeast. Using locus-specific optogenetic recruitment and live-cell microscopy, we measure dynamic expression profiles within single cells. CAP recruitment elicits a range of responses varying in activation delay, strength, production rate, and noise. We find that promoter activity is characterized by graded, rather than switch-like, transitions. A kinetic model with three promoter states and a positive feedback loop successfully captures the key features of expression driven by each CAP. These results reveal the rich dynamic landscape possible from a single gene, offering insights into native cellular processes and enhancing gene expression control in synthetic biology.
|
|
Scooped by
mhryu@live.com
May 5, 3:43 PM
|
IQ-TREE (https://iqtree.github.io/) is a widely used open-source software tool for efficiently inferring phylogenetic trees under maximum likelihood. Here, we present IQ-TREE version 3, the third major release of the software. IQ-TREE 3 significantly extends version 2 with new features, including mixture models as an alternative to partitioned models, gene and site concordance factors to quantify discordance between genomic regions, integration with phylogenomic divergence time estimation, and a fully-featured sequence simulator. The IQ-TREE 3 source code is available at https://github.com/iqtree/iqtree3.
|






















m-1str, industry