 Your new post is loading...

|
Scooped by
mhryu@live.com
Today, 11:02 AM
|
Certain peptoids designed as mimics of host defense peptides such as LL-37 exhibit potent, broad-spectrum antibacterial, antifungal, antiparasitic, and antiviral activity with minimal cytotoxicity. Previous fixed-cell studies have suggested that the peptoids can pass through bacterial membranes and rapidly kill bacteria by aggregating intracellular macroanions, including ribosomes and DNA. However, the dynamic mechanisms of action of these biomimetic peptoids have remained elusive. We employed single-bacterial-cell, time-resolved fluorescence microscopy, and single-particle tracking methods to investigate the effects of the 12mer peptoid TM1, along with shorter alkylated and brominated analogues, on cytoplasmic membrane permeabilization and DNA and ribosome rigidification of Escherichia coli. Our results demonstrate that TM1 and several of its analogues permeabilize the cytoplasmic membrane within five minutes of flowing the peptoid solution over the cells—faster than seen for the important human antimicrobial peptide LL-37—and rigidify DNA and ribosomes as effectively as LL-37. Detailed biophysical structural and dynamical studies show that TM1 binds to both DNA (double-stranded and single-stranded) and single-stranded RNA in a similar manner to LL-37, which is well known to display strong nucleic acid binding. These results support our hypothesis that TM1 and its analogues exert their antimicrobial effects through intracellular aggregation of biomacromolecules such as ribosomes, RNA, and DNA. TM1 displays a higher affinity for RNA compared to DNA, suggesting it will preferentially bind in vivo to bacterial ribosomes. Our study yields insight into the dynamic effects of antimicrobial peptoids, facilitating their future development as biomimetic anti-infectives, with the additional advantage of protease invulnerability.
|
|
Scooped by
mhryu@live.com
Today, 10:36 AM
|
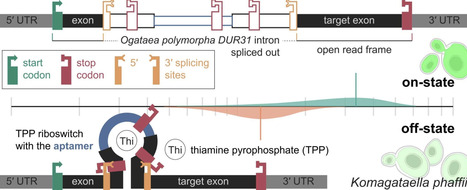
Komagataella phaffii (syn. Pichia pastoris) is a methylotrophic yeast widely established as host for recombinant protein production and increasingly used as cell factory for C1-based metabolites. Nevertheless, synthetic biology tools for regulating protein expression in this host remain limited, relying largely on promoters of genes from central carbon metabolism. Riboswitches, i.e. mRNA elements that regulate translation or splicing via ligand-dependent folding, offer a complementary layer of regulation, yet remain unused in K. phaffii synthetic biology. In the methylotrophic yeast Ogataea polymorpha, an intron within the DUR31 gene acts as a thiamine pyrophosphate (TPP) riboswitch, downregulating gene expression through alternative splicing, and retaining a premature stop codon in response to exogenous thiamine. We evaluated this TPP riboswitch in K. phaffii as a tool to modulate expression from the glycolytic GAP promoter. Using EGFP and destabilised UBIYΔkGFP⁎ as reporters in combination with flow cytometry, we show that the orthogonal riboswitch is functional in K. phaffii. Like many other described riboswitches, the system exhibits substantial basal (leaky) expression and cannot be considered a tight molecular on/off switch. RT-PCR confirmed the presence of both spliced and unspliced transcripts in the cells regardless of external thiamine supplementation. Unexpectedly, inserting an additional exon upstream of the riboswitch intron abolished detectable downstream protein production, indicating that splicing efficiency is strongly influenced by the local sequence context of the splice sites.
|
|
Scooped by
mhryu@live.com
Today, 2:01 AM
|
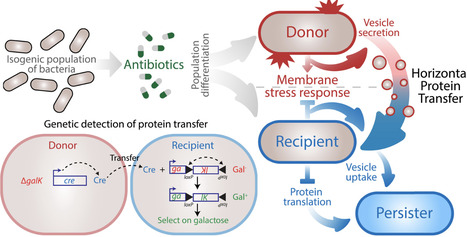
The exchange of biological matter between bacterial cells drives adaptation and evolution. However, whether bacteria can exchange functional proteins remains unclear. In this work, we found that antibiotic treatment can induce vesicle-mediated horizontal protein transfer within and between bacterial species. We developed a genetic system in E. coli to track transfer events and performed single-cell transcriptomic profiling on an isogenic population of bacteria. Antibiotics stimulated the differentiation of this isogenic population into distinct cell states: donor cells that activated a membrane stress response to release protein-containing vesicles and recipient cells that suppressed this response to acquire protein from their neighbors. Protein uptake enhanced the antibiotic persistence of recipient cells, revealing that vesicle exchange promotes bacterial survival during antibiotic treatment.
|
|
Scooped by
mhryu@live.com
Today, 1:45 AM
|
Telomeres are sequences at chromosome ends that distinguish the natural end from a DNA break. Telomeres shorten at each round of cell division because the replisome cannot completely copy both DNA strands to the very end. This shortening is counterbalanced by telomerase, which adds telomeric sequences de novo onto telomeres. The balance of shortening and lengthening is regulated to establish an equilibrium distribution of telomere lengths. If the equilibrium is perturbed, short telomeres trigger a DNA damage response that leads to cellular senescence or cell death. In humans, short telomeres cause age-related degenerative disease, while long telomeres protect against senescence and allow the continued growth of cancer cells. To target the telomere in human disease, we need a more complete understanding of how telomere length is regulated. Here, we describe pathways that regulate telomere length with a focus on the fundamental mechanisms established over the last 40 years in the yeast Saccharomyces cerevisiae.
|
|
Scooped by
mhryu@live.com
Today, 1:25 AM
|
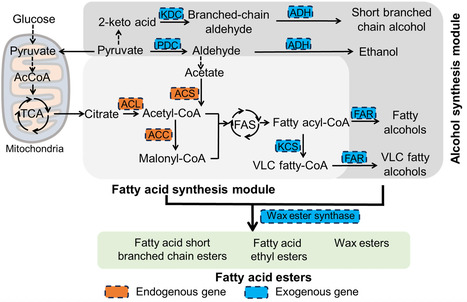
Fatty acid esters are building blocks in next-generation biofuels, eco-friendly detergents, crop-enhancing adjuvants, and high-value cosmetic emollients. However, their industrial production relies on two unsustainable approaches: petrochemical-derived chemical catalysis that poses sustainability challenges and botanical extraction processes that exacerbates land-use conflicts. Here we develop a de novo microbial biosynthesis platform using the non-conventional oleaginous yeast Rhodotorula toruloides as a chassis, and achieve high-level biosynthesis of structurally diversified esters without the addition of alcohol or lipid precursors. By screening specific pathway enzymes and conducting modular pathway engineering, we successfully reprogram the native lipogenic metabolism of R. toruloides for de novo synthesis of fatty acid ethyl esters (FAEEs) at 579 mg/L, fatty acid short-branched chain esters (FASBEs) at 169 mg/L, and wax esters (WEs) at 1.30 g/L in shake-flask fermentation. As a case study, we optimize the synthesis of WEs in a 5 L fermenter, and achieve a production of 13.04 g/L. These engineered strains potentially offer an efficient, economical and environmentally friendly platform for the industrial production of fatty acid esters and oleochemicals. Fatty acid esters are biofuels, eco-friendly detergents, and cosmetic emollients, however, their industrial production is unsustainable. Here the authors develop a de novo biosynthesis platform using the yeast Rhodotorula toruloides and achieve high-level production of structurally diverse esters.
|
|
Scooped by
mhryu@live.com
Today, 1:16 AM
|
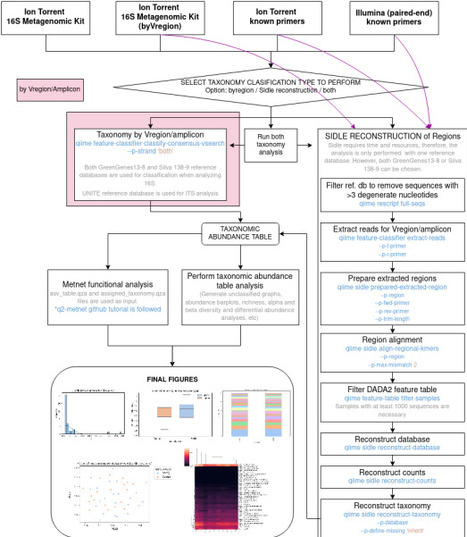
Microbiome analysis has become a pivotal tool in understanding the role of microbiota in human health and disease. However, the lack of standardized workflows, together with the limitations of proprietary software solutions, hampers reproducibility and flexibility. Here, we present Mbiome, an open-source, user-friendly and automated workflow designed to streamline amplicon-based microbiome analysis. Built upon QIIME2, Mbiome supports both bacterial (16S rRNA) and fungal (ITS) profiling, and is compatible with raw fastq files generated by Ion Torrent (IT) and Illumina (IL) sequencing platforms. The workflow guides users through an interactive setup process via a simple configuration file, enabling researchers with minimal bioinformatics experience to perform comprehensive analyses without writing code. Once configured, Mbiome automates major steps including quality control, taxonomic assignment, α and β-diversity analyses, functional predictions (via q2-metnet), and customizable visualizations and statistical analyses. Mbiome has been validated using real-world datasets from multiple sclerosis research projects, performing a comparison between different microbiome analysis approaches, including 16S hypervariable region reconstruction, amplicon-based strategies, and cross-platform sequencing (IT and IL), as well as against results obtained with Ion Reporter (IR) commercial software. This evaluation demonstrated its versatility and effectiveness across different sequencing platforms. Moreover, Mbiome provided improved flexibility, transparency, and taxonomic resolution compared to IR. By combining accessibility, reproducibility, and cross-platform compatibility, Mbiome lowers the barrier to microbiome data analysis and facilitates high-quality, standardized workflows in both research and applied settings. Mbiome is publicly available at https://github.com/MGorostidi/mbiome.
|
|
Scooped by
mhryu@live.com
Today, 1:10 AM
|
Rhodopseudomonas palustris TIE-1 (TIE-1) is a metabolically versatile environmental bacterium that flourishes across gradients of iron, oxygen, and light. This versatility necessitates extensive regulatory control, exemplified by the aerobic-anaerobic metabolic shift controlled by the hierarchy of CRP/FNR-family regulators AadR and FixK. Many anaerobic metabolic pathways demand expression of iron cofactor-intensive proteins, and TIE-1 in particular can generate energy through phototrophic iron oxidation via the PioABC system. However, TIE-1 lacks canonical iron-sensing regulators: IscR, ancestral Fe(II)-sensing Fur, and Fe(II)-sensing RirA of Rhizobiaceae, leaving it unclear how TIE-1 coordinates expression of these iron-requiring metabolisms with bioavailable iron levels. Here, we demonstrate that the AadR-FixK hierarchy plays a previously underappreciated role in iron regulation in TIE-1 by comparing growth and transcription in wild-type and regulatory mutants across wetland-inspired naturomimetic conditions. ΔaadR and ΔfixK showed defects in iron-dependent growth and Fe(II) oxidation, and the ΔaadRΔfixK double mutant was synthetically lethal under anaerobiosis. The regulatory hierarchy of FixK and AadR influences expression of Fur-family regulators: the two irr paralogs were oppositely regulated in the presence of AadR, and absence of AadR perturbed iron-responsive expression of mur. Furthermore, the AadR regulon was significantly enriched for iron-related and iron-containing proteins. Despite initial predictions that AadR directly regulates pioABC, we found no conclusive evidence for direct AadR activity at the pioABC promoter, refining the search for pio regulators. Together, these findings establish AadR as a central integrator of oxygen and iron signals to coordinate iron-requiring anaerobic metabolism in TIE-1.
|
|
Scooped by
mhryu@live.com
Today, 12:59 AM
|
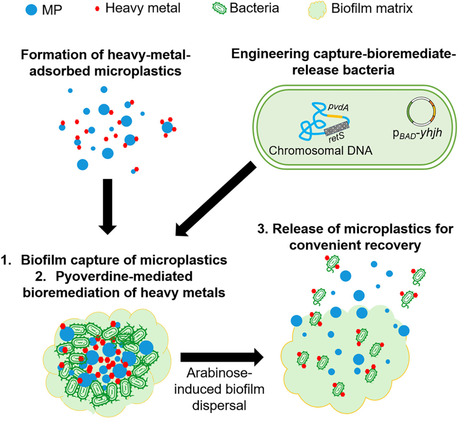
Microplastics (MPs) pose escalating environmental and human health risks, particularly when they adsorb heavy metals and form complex, co-contaminated pollutants. Furthermore, when MPs are removed and sent to landfills or incineration, the adsorbed heavy metals often persist and re-enter the environment. However, most existing remediation strategies remove only single pollutants, which fail to address complex pollution. In this research article, we engineered an environmental Pseudomonas aeruginosa strain with three programmable functions: (i) enhanced biofilm formation for efficient aggregation and capture of MPs, (ii) increased pyoverdine production for bioremediation of heavy metals (lead and cadmium) adsorbed on MPs, and (iii) subsequent arabinose-inducible biofilm dispersal to release decontaminated MPs for convenient recovery. Our proof of concept was validated in a pilot trial with polluted Hong Kong seawater, demonstrating efficient dual-pollutant removal in environmentally relevant conditions. Hence, our work highlights the promise of biotechnology in advancing multi-pollutant environmental remediation.
|
|
Scooped by
mhryu@live.com
Today, 12:15 AM
|
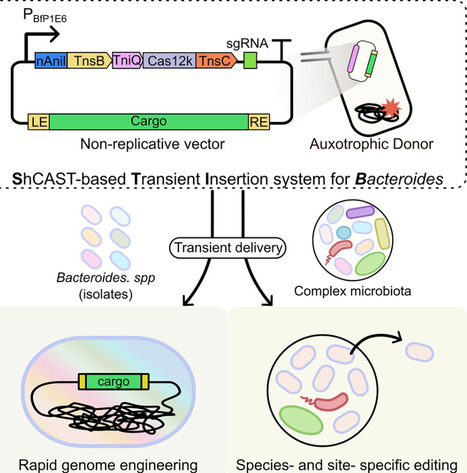
Gut Bacteroides are abundant and critical to human health, yet most are genetically cumbersome, non-model microbes. A widely applicable editing tool for Bacteroides is essential for gut microbiome manipulation. Here, we develop STIB (ShCAST-based transient insertion system for Bacteroides), an efficient genome-editing tool derived from CRISPR-associated transposases that enables rapid and site-specific insertions independent of homologous recombination. By fusing a nicking homing endonuclease to the transposase and an ATPase to Cas12k, we systematically optimize STIB to minimize plasmid cointegration and achieve >97% on-target insertion. STIB exhibits broad applicability across different genomic loci in diverse Bacteroides species, including non-model species. Finally, we apply STIB to achieve species- and site-specific editing of distinct Bacteroides species within a complex synthetic gut microbiota. Overall, STIB expands the toolbox for the functional investigation and engineering of the human microbiome.
|
|
Scooped by
mhryu@live.com
June 29, 11:52 PM
|
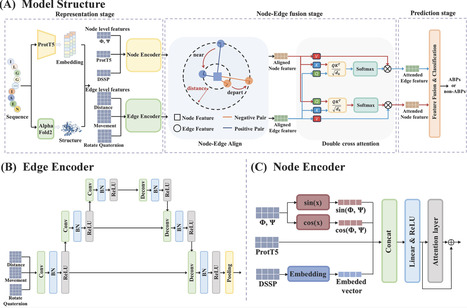
Antibacterial peptides (ABPs) represent a promising alternative strategy for combating antimicrobial resistance. Graph neural networks and their variants have shown substantial potential in ABP identification. However, most existing graph-based methods focus on node features while neglecting edge features. Edge features play an important role in characterizing peptide conformation and spatial stability, thereby directly influencing interactions with bacterial membranes and antibacterial activity. Moreover, node and edge features extracted by heterogeneous encoders reside in disparate feature spaces, making their effective integration nontrivial. These challenges highlight the need for an explicit alignment mechanism to bridge heterogeneous representations. In this study, we propose CLABP, a contrastive learning-based framework for ABP identification that integrates edge features. Specifically, backbone dihedral angles, ProtT5 embeddings, and Define Secondary Structure of Proteins-derived secondary structures were employed as node features, while inter-residue distance, motion vectors, and rotation quaternions were used to construct edge features. Node and edge representations were independently extracted using dedicated encoders and subsequently aligned into a shared latent space via contrastive learning, which minimizes the distance between paired representations derived from the same peptide. The aligned features were then fused through a dual cross-attention mechanism for downstream prediction. CLABP outperforms other state-of-the-art methods, achieving an accuracy of 92.6% and a Matthews correlation coefficient of 0.853. Ablation studies further confirm that both edge features and the alignment mechanism are critical to model performance.
|
|
Scooped by
mhryu@live.com
June 29, 11:32 PM
|
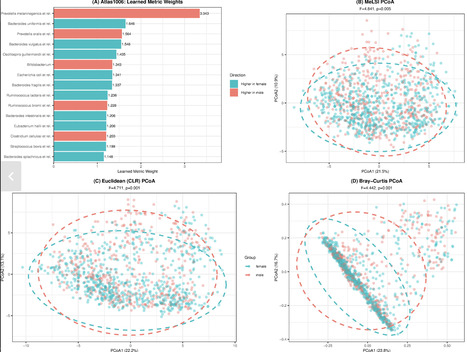
Microbiome beta diversity analysis relies on distance-based methods, including permutational multivariate analysis of variance (PERMANOVA) combined with fixed ecological distance metrics (Bray-Curtis, Euclidean, Jaccard, and UniFrac), which treat all microbial taxa uniformly, regardless of their biological relevance to community differences. This “one-size-fits-all” approach may miss subtle but biologically meaningful patterns in complex microbiome data. We present Metric Learning for Statistical Inference (MeLSI), a novel machine learning framework that learns data-adaptive distance metrics optimized for detecting community composition differences in multivariate microbiome analyses. MeLSI employs an ensemble of weak learners using bootstrap sampling, feature subsampling, and gradient-based optimization to learn optimal feature weights, combined with rigorous permutation testing for statistical inference. The learned metrics can be used with PERMANOVA for hypothesis testing and with principal coordinates analysis for ordination visualization. Comprehensive validation on synthetic benchmarks and real data sets shows that MeLSI maintains proper type I error control while delivering competitive or superior statistical power for detecting subtle community shifts and, crucially, supplies interpretable feature-weight profiles that clarify which taxa drive group separation. On the DietSwap data set, MeLSI was the only method to achieve significance at α = 0.05, demonstrating that adaptive weighting can detect diet-induced community shifts that fixed metrics miss. Across all data sets, the learned feature weights identified biologically relevant taxa while providing actionable insight that no fixed distance metric can supply. MeLSI therefore offers a statistically rigorous tool that augments beta diversity analysis with transparent, data-driven interpretability.
|
|
Scooped by
mhryu@live.com
June 29, 11:18 PM
|
The skin microbiome maintains cutaneous homeostasis through colonization resistance and immune modulation, targeted prebiotic interventions however remain largely unexplored. The present study addresses this lacuna effectively demonstrating the ability of plant-derived prebiotics to selectively modulate key skin commensals and pathogenic bacteria thereby unraveling a new dimension to use of prebiotics in skin care. Linum usitatissimum (flaxseed) and Allium sativum (garlic) extracts used herein exhibited complete growth inhibition of Staphylococcus aureus within 6 h, while Curcuma amada (mango ginger) rapidly halted the growth of Cutibacterium acnes within 15 min. Gas chromatography-mass spectrometry was carried out to unveil the bioactive constituents in plant-prebiotics as well as the exposed organisms. Field emission gun-scanning electron microscopy validated bacterial cell membrane disruption correlating with antimicrobial efficacy. Remarkably, Allium cepa (onion) and Tinospora cordifolia (guduchi) selectively enhanced the proliferation of Staphylococcus epidermidis while simultaneously inhibiting pathogenic species. Metabolomic profiling revealed that prebiotic-stimulated S.epidermidis produced elevated levels of butyric and succinic acids that are documented to have anti-bacterial activity. Plant-based prebiotics can thus be strategically reinforced to benefit skin microbiota with simultaneous inhibition of skin pathogens, providing a scientific foundation for microbiome-targeted cosmeceuticals.
|
|
Scooped by
mhryu@live.com
June 29, 11:01 PM
|
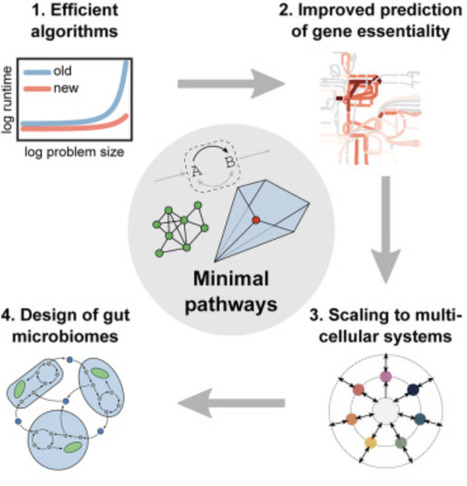
Many interactions in microbial consortia or tissues of multicellular organisms rely on networks of metabolite exchanges. To predict community function and composition beyond statistical correlations, one can use genome-scale metabolic models. However, comprehensive model analysis via metabolic pathways is a major challenge because pathway counts grow combinatorially with model size. Here, we define minimal pathways that yield compact representations of metabolic network capabilities. They generalize existing pathway concepts by allowing inhomogeneous constraints and targeted analysis of subnetworks, and we show how to enumerate and sample them efficiently via iterative minimization and pathway graphs. This enables applications such as assessing quantitative gene essentiality in the central metabolism of E. coli, predicting metabolite exchanges associated with homeostasis and health in a host-microbe model of the human gut, and designing butyrate-producing microbial communities. Minimal pathways enable scalable analysis of metabolic subnetworks such as metabolite exchanges in uni- and multicellular systems.
|
|
|
Scooped by
mhryu@live.com
Today, 10:50 AM
|
Despite the groundbreaking advances in deep learning-enabled methods for biomolecular modeling, predicting accurate three-dimensional (3D) structures of RNA remains challenging owing to the highly flexible nature of RNA molecules combined with the limited availability of evolutionary sequences or structural homology. Here we introduce RNAbpFlow, a sequence- and base pair-conditioned SE(3)-equivariant flow-matching model for generating RNA 3D structural ensembles. Leveraging a nucleobase center representation, RNAbpFlow enables end-to-end generation of all-atom RNA structures without the explicit or implicit use of evolutionary information or homologous structural templates. Experimental results show that base-pairing conditioning leads to broadly generalizable performance improvements over current approaches for RNA topology sampling and predictive modeling in large-scale benchmarking. RNAbpFlow generates all-atom RNA conformational ensembles for single-chain RNA monomers without the explicit or implicit use of evolutionary information or homologous structural templates.
|
|
Scooped by
mhryu@live.com
Today, 2:11 AM
|
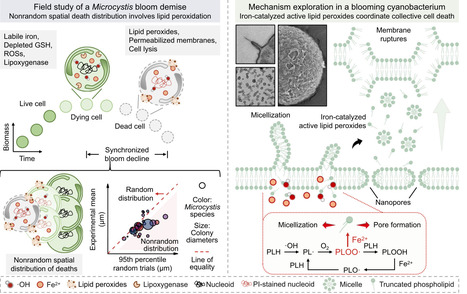
Harmful algal blooms, the most severe ecological hazards worldwide, terminate abruptly within a few days. In this work, we identified that iron-catalyzed active lipid peroxides predominantly trigger individual cell ferroptosis and drive the population collapse of blooming cyanobacteria. We reveal the chronological sequence of labile iron burst, oxidative stress, lipid peroxidation, and cell death during a Microcystis bloom demise event. Dead cells exhibit a nonrandom spatial distribution within colonies. Intensifying lipid peroxidation catalyzed by cellular labile iron generates truncated phospholipids with shortened fatty acyl chains bearing alkyl groups. These active lipid peroxides destabilize plasma membranes and induce nanoscale membrane pore formation, resulting in individual cell ferroptosis and lysis. Oxidized lipids are also released from ferroptotic cells, propagating lipid peroxidation to neighboring cells, thereby spreading death throughout the population.
|
|
Scooped by
mhryu@live.com
Today, 1:50 AM
|
Bacteria regulate homeostatic growth by adjusting proteome composition. In E. coli, this coordination is mediated by guanosine tetraphosphate and pentaphosphate, collectively termed (p)ppGpp, which couple amino acid supply with ribsosome production. We identified a distinct architecture in Bacillus subtilis, in which guanosine triphosphate (GTP), not (p)ppGpp, controls proteome allocation. Translational inhibition resulted in GTP depletion and suppressed amino acid biosynthesis through feedback inhibition without altering ribosome abundance, establishing a regulated decoupling between total amino acid flux and proteome composition, with flux deviating from proteome-based predictions. By artificially adjusting GTP concentrations, we recoupled flux and proteome, restoring growth to maximal amounts. The regulated suboptimality enables a trade-off to balance growth and stress resilience. Similar GTP-based strategies were present in other Firmicute species, indicating possible evolutionary conservation. Proteome composition and metabolic flux have distinct regulatory layers in some bacteria.
|
|
Scooped by
mhryu@live.com
Today, 1:42 AM
|
Single cell protein (SCP), distinguished by its high production efficiency, flexible substrate utilization, and excellent nutritional value, has broad application prospects in both feed and food sectors. With the advancement of synthetic biology and metabolic engineering, SCP is poised to become a mainstream protein source, providing key solutions for global food security and carbon neutrality goals. This review focuses on Saccharomyces cerevisiae as an SCP chassis and summarizes recent metabolic and synthetic biology strategies aimed at enhancing cellular protein content and cell biomass, thereby improving its potential for sustainable large-scale protein production.
|
|
Scooped by
mhryu@live.com
Today, 1:20 AM
|
Protein engineering often relies on separate models for related developability properties, limiting efficiency and transfer across tasks. We present Prot2Prop, a multitask framework based on a frozen ProstT5 encoder with shared and task-specific adapters for joint prediction of six protein properties: material production, solubility, temperature stability, aggregation propensity, expression yield, and folding stability. Across held-out test data, Prot2Prop achieved strong performance on both classification and regression tasks, including AUROC values ranging from 0.86 to 0.98 for classification endpoints and Spearman correlations ranging from 0.73 to 0.86 for regression endpoints. The model achieved particularly strong performance for temperature stability (AUROC = 0.98) and aggregation propensity (Spearman = 0.86). Post-hoc calibration further improved regression accuracy, reducing folding stability MAE from 0.67 to 0.48. These results demonstrate that parameter-efficient multitask adaptation of protein language models can provide accurate and unified prediction of diverse protein developability properties.
|
|
Scooped by
mhryu@live.com
Today, 1:13 AM
|
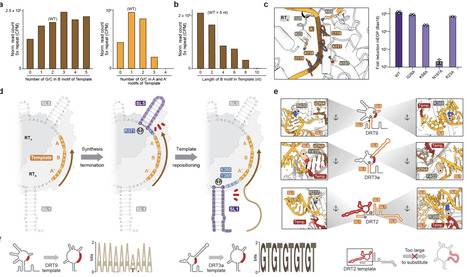
Defense-associated reverse transcriptases (DRTs) employ DNA synthesis to protect bacteria against phage infection. We previously showed that DRT10, a tripartite system comprising an RT, a noncoding RNA (ncRNA), and a SLATT effector protein, catalyzes protein-primed, tandem-repeat DNA synthesis in a mechanism strikingly analogous to eukaryotic telomerase. However, the structural basis by which the RT-ncRNA complex directs repeat addition processivity and controls repeat length remains unknown. Here we present cryo-EM reconstructions of two evolutionarily diverse DRT10 RT-ncRNA systems that reveal an unanticipated 2:1 architecture, wherein two RT monomers bind opposite sides of a single, pseudo-symmetric ncRNA. Biochemical experiments demonstrate that each RT monomer reverse transcribes the template encoded on its respective side of the ncRNA, but only one generates the long repetitive product, with the template sequence defined by the distance between two flanking stem-loop anchors. Together with earlier studies of DRT2, DRT3, and DRT9, our findings identify a conserved mechanistic logic underlying ncRNA-templated tandem-repeat synthesis across Class 2 UG antiviral systems, despite vastly different architectural solutions.
|
|
Scooped by
mhryu@live.com
Today, 1:05 AM
|
Serpentinization produces hyperalkaline, H₂- and CH₄-rich fluids that support microbial life and serve as analogs for ocean worlds such as Enceladus. While methane production in these systems has been well studied, methane consumption—especially under high pH—remains poorly understood. Here, we present isotopic, geochemical, and genomic evidence for hyperalkaliphilic (pH > 11) methanotrophy in the Samail ophiolite of Oman. Using models that account for fluid mixing and gas exsolution, we identify δ¹³CH₄ enrichment that cannot be explained by abiotic processes alone. The enrichment of 13CH4 co-occurs with methanotroph 16S rRNA gene sequences, particularly in fluids formed by mixing CH₄-rich, reduced fluids with oxidant-rich waters. Shotgun metagenome sequencing reveals a metagenome-assembled genome affiliated with Methylovulum, encoding a complete methane oxidation pathway, multiple carbon assimilation routes, and Na⁺/H⁺ antiporters—adaptations likely enabling growth above pH 11. Our findings highlight the viability of methanotrophy under extreme high pH conditions and provide a framework for interpreting δ¹³CH₄ signals in serpentinizing environments on Earth and beyond. Serpentinization makes H₂- and CH₂-rich fluids analogous to ocean worlds like Enceladus. Isotopes, chemistry, and genomes show methane consumption at pH >11 in Oman—revealing extreme methanotrophy on Earth and insights into CH4 as a biosignature.
|
|
Scooped by
mhryu@live.com
Today, 12:43 AM
|
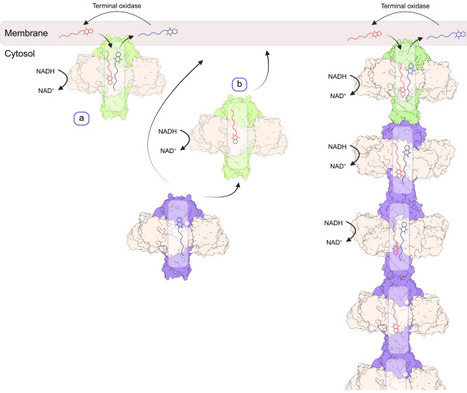
Cellular respiration depends on transferring electrons to hydrophobic quinones in membrane bilayers, constraining capacity to available surface area. Expanding this capacity is thought to have driven cellular complexity and eukaryogenesis, with Gram-negative bacteria evolving internal invaginations and eukaryotes using membrane-bound organelles. Whether Gram-positive bacteria, which lack such membranes, evolved alternatives was unknown. Here, we show that Bacillus subtilis forms a quinone-transporting pseudomembrane composed of filaments of the NADH dehydrogenase Ndh and the quinone-transporting protein Ncp. Cryo-EM, lipidomics, and molecular dynamics reveal that Ndh and Ncp co-assemble with phospholipids into a complex containing a solvent-excluded hydrophobic lumen that sequesters quinones. These complexes further assemble into filaments, linking chambers into a continuous conduit that amplifies quinone reduction while occupying minimal membrane space. Phylogenetic analysis suggests this recent innovation is widespread in Bacillota. Quinone-transporting filaments thus reveal a third strategy for overcoming surface-area limits and provide principles for engineering synthetic energy systems.
|
|
Scooped by
mhryu@live.com
Today, 12:05 AM
|
Despite progress in automated gene annotation, many deficiencies and knowledge gaps remain, even for well-studied organisms. Of particular concern is the accuracy and detail of annotations for transporters of various organic substrates and products of metabolism and for enzymes that do not share sequence homology with well-characterized strains. Unfortunately, annotation errors present in earlier genome-scale metabolic (GSM) models propagate to newer models with few opportunities for later correction. Here, we introduce a systematic computational procedure that applies the E. coli genome-scale metabolic model iML1515, extended with transcriptional regulatory rules, to design auxotrophs that can grow on glucose but fail to grow on different carbon substrate(s) unless rescued with the addition of an ORF encoding a complementation metabolic function (transport and enzymatic reactions). Using the E. coli GSM model supplemented with regulatory rules that quantify growth/no growth outcomes on different organic substrates, we identified 258 distinct auxotrophic designs (97 single-gene, 142 double-gene, and 19 triple-gene knockouts) for which specific single functions can uniquely complement them. Experimental validation of 61 single-knockout strains demonstrated 59% confirmed auxotrophy and 28% partial auxotrophy. We envision that this collection of auxotrophic strains can be used to disambiguate the metabolic role of unannotated or poorly annotated genes.
|
|
Scooped by
mhryu@live.com
June 29, 11:44 PM
|
CRISPR spacer-protospacer matching is widely used to infer host-virus interactions in microbial and viromics studies, but the choice of sequence search or alignment tool and its reporting behavior is often under-evaluated for this specific task. Using synthetic, semi-synthetic, and real datasets, we benchmarked commonly used tools and observed substantial differences in recall, runtime, and resource usage across distance metrics and thresholds. Our analyses support practical defaults for large-scale spacer-target matching and clarify trade-offs between exhaustive and heuristic approaches.
|
|
Scooped by
mhryu@live.com
June 29, 11:25 PM
|
Lignocellulose is a major component of plant biomass and is recalcitrant, with efficient degradation typically requiring oxygen-dependent oxidative and carbohydrate-active enzymes (CAZymes). Anaerobic turnover is slower but can be supported by microbes capable of nitrate respiration, including denitrifiers and dissimilatory nitrate reduction to ammonium (DNRA) bacteria, which may use nitrate or nitric oxide as alternative oxidants. Anoxic layers beneath the oxic zones of eutrophic lake sediments, where nitrate penetrates from surface waters, provide a natural habitat for such organisms. To investigate these processes, we established nitrate-amended enrichments from organic-rich sediments of 10 eutrophic lakes and applied gas kinetics alongside metagenomics and metaproteomics to characterize the microbial communities. We identified a set of core microbial metagenome-assembled genomes (MAGs) present in all enrichments, dominated by Pseudomonadota, Bacteroidota, Verrucomicrobiota, and Actinomycetota, which played key roles in denitrification and fermentation. Lignocellulose degradation, however, was largely carried out by species outside the core microbiome—that is, different key degraders between lakes, suggesting lake-specific specialization. Among these, we observed potential respiratory DNRA pathways and a broad repertoire of CAZymes targeting various lignocellulose subfractions. Interestingly, many MAGs also encoded nitric oxide dismutases (NODs), enzymes postulated to convert NO to molecular oxygen and dinitrogen gas. Together, these findings advance our understanding of anaerobic biomass degradation and nitrogen cycling in eutrophic freshwater sediments, while highlighting the unexplored functional diversity of NOD-containing bacteria as an intriguing open question for future research.
|
|
Scooped by
mhryu@live.com
June 29, 11:15 PM
|
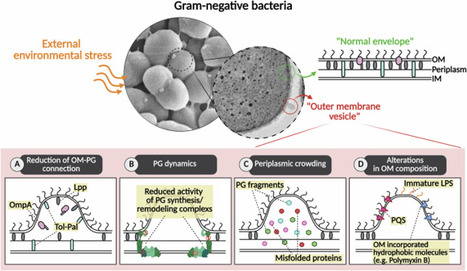
Nano-sized outer membrane vesicles (OMV) are lipid-bilayered structures that primarily encapsulate periplasmic components, with minor inclusion of cytoplasmic materials. Rather than passive byproducts of cellular damage, OMVs are now understood as active mediators of bacterial physiology, environmental adaptation, and host interaction. Recent evidence identifies envelope instability as a key mechanistic driver of OMV biogenesis. Disruptions in outer membrane–peptidoglycan connectivity, imbalances in periplasmic homeostasis, and alterations in lipid asymmetry collectively promote vesicle formation as a regulated adaptive response. Under stress conditions, OMVs acquire specialized functional roles, selectively enriching specific cargo and exhibiting surface properties that enable them to sequester host-derived antimicrobial factors. OMV-associated biomolecules further influence vesicle uptake into host cells through distinct endocytic pathways, shaping intracellular trafficking and downstream functional outcomes. Following internalization, pathogen-derived OMVs disrupt host signaling pathways and are exploited to promote immune evasion, whereas commensal-derived OMVs contribute to microbiota homeostasis and immune modulation. In parallel, growing efforts to harness OMVs as vaccine platforms highlight their potential as innovative tools for both therapeutic and prophylactic applications. Collectively, these insights position OMVs as critical mediators of bacterial pathogenicity and as promising targets for anti-infective strategies. A review summarizes recent insights into outer membrane vesicles (OMVs) in Gram negative bacteria, including their biogenesis mechanisms, stress-associated cargo remodeling, host-cell interactions, immunomodulatory roles, and therapeutic applications.
|


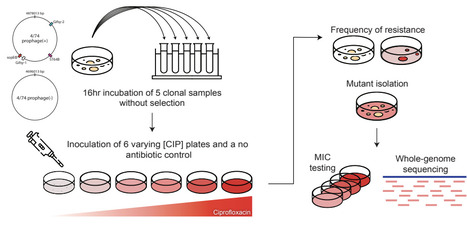

















Experimental workflow for isolation of CIP-resistant mutants. Five independent clonal replicates of 4/74 prophage(+) and 4/74 prophage(-) were grown for 16 h in antibiotic-free liquid culture to avoid prophage induction, then plated on CIP gradient plates and antibiotic-free controls. After incubation, surviving colonies were counted and isolated for downstream phenotypic and genotypic analyses.
resistant lysogens are rarer but more resistant means: among bacteria that carry prophages and were exposed to ciprofloxacin, fewer survived selection compared to prophage-free bacteria