



Follow, research and publish the best content
Get Started for FREE
Sign up with Facebook Sign up with X
I don't have a Facebook or a X account
Already have an account: Login

Publications from The Sainsbury Laboratory
66.8K views |
+2 today
 Your new post is loading...
Your new post is loading... Your new post is loading...
Your new post is loading...
The Sainsbury Lab's insight:
Plants have evolved strong innate immunity mechanisms, but successful pathogens evade or suppress plant immunity via effectors delivered into the plant cell. Hyaloperonospora arabidopsidis (Hpa) causes downy mildew on Arabidopsis thaliana, and a genome sequence is available for isolate Emoy2. Here, we exploit the availability of genome sequences for Hpa and Arabidopsis to measure gene-expression changes in both Hpa and Arabidopsis simultaneously during infection. Using a high-throughput cDNA tag sequencing method, we reveal expression patterns of Hpa predicted effectors and Arabidopsis genes in compatible and incompatible interactions, and promoter elements associated with Hpa genes expressed during infection. By resequencing Hpa isolate Waco9, we found it evades Arabidopsis resistance gene RPP1through deletion of the cognate recognized effector ATR1. Arabidopsis salicylic acid (SA)-responsive genes including PR1 were activated not only at early time points in the incompatible interaction but also at late time points in the compatible interaction. By histochemical analysis, we found that Hpa suppresses SA-inducible PR1 expression, specifically in the haustoriated cells into which host-translocated effectors are delivered, but not in non-haustoriated adjacent cells. Finally, we found a highly-expressed Hpa effector candidate that suppresses responsiveness to SA. As this approach can be easily applied to host-pathogen interactions for which both host and pathogen genome sequences are available, this work opens the door towards transcriptome studies in infection biology that should help unravel pathogen infection strategies and the mechanisms by which host defense responses are overcome. 
The Sainsbury Lab's insight:
Plant growth and development depend on the biosynthesis and remodeling of the cell wall. To coordinate these two processes, surveillance mechanisms have evolved to monitor the state of the cell wall. The brassinosteroid (BR) hormone signaling pathway plays an essential role in growth control and regulates the expression of a plethora of cell wall-related genes. We have previously shown that feedback signaling from the wall can modulate the outputs of the BR pathway, ensuring cell wall homeostasis and integrity. Here, we identified a receptor-like protein (RLP44), which mediates the activation of BR signaling through direct interaction with the BR coreceptor BAK1. Thus, RLP44 integrates cell wall surveillance with hormone signaling to control cell wall integrity and growth.
The Sainsbury Lab's insight:
The CRISPR/Cas9 system is highly efficient at generating targeted mutations in stable transgenic tomato plants, and homozygous deletions of a desired size can be created in the first generation.
The Sainsbury Lab's insight:
Pathogens can colonize all plant organs and tissues. To prevent this, each cell must be capable of autonomously triggering defence. Therefore, it is generally assumed that primary sensors of the immune system are constitutively present. One major primary sensor against bacterial infection is the FLAGELLIN SENSING 2 (FLS2) pattern recognition receptor (PRR). To gain insights into its expression pattern, the FLS2 promoter activity in β-glucuronidase (GUS) reporter lines was monitored. The data show that pFLS2::GUS activity is highest in cells and tissues vulnerable to bacterial entry and colonization, such as stomata, hydathodes, and lateral roots. GUS activity is also high in the vasculature and, by monitoring Ca2+responses in the vasculature, it was found that this tissue contributes to flg22-induced Ca2+ burst. The FLS2 promoter is also regulated in a tissue- and cell type-specific manner and is responsive to hormones, damage, and biotic stresses. This results in stimulus-dependent expansion of the FLS2 expression domain. In summary, a tissue- and cell type-specific map of FLS2 expression has been created correlating with prominent entry sites and target tissues of plant bacterial pathogens.

Reference-free SNP detection, that is identifying SNPs between samples directly from comparison of primary sequencing data with other primary sequencing data and not to a pre-assembled reference genome is an emergent and potentially disruptive technology that is beginning to open up new vistas in variant identification that reveals new applications in non-model organisms and metagenomics. The modern, effcient data structures these tools use enables researchers with a reference sequence to sample many more individuals with lower computing storage and processing overhead. In this article we will discuss the technologies and tools implementing reference-free SNP detection and the potential impact on studies of genetic variation in model and non-model organisms, metagenomics and personal genomics and medicine.
The Sainsbury Lab's insight:
Rapid technological advances have led to an explosion of biomedical data in recent years. The pace of change has inspired new, collaborative approaches for sharing materials and resources to help train life scientists both in the use of cutting-edge bioinformatics tools and databases, and in how to analyse and interpret large datasets. A prototype platform for sharing such training resources was recently created by the Bioinformatics Training Network (BTN). Building on this work, we have created a centralised portal for sharing training materials and courses, including a catalogue of trainers and course organisers, and an announcement service for training events. For course organisers, the portal provides opportunities to promote their training events; for trainers, the portal offers an environment for sharing materials, for gaining visibility for their work and promoting their skills; for trainees, it offers a convenient one-stop shop for finding suitable training resources and identifying relevant training events and activities locally and world-wide.
EMBO Journal: Negative control of BAK1 by protein phosphatase 2A during plant innate immunity (2014)
The Sainsbury Lab's insight:
Recognition of pathogen‐associated molecular patterns (PAMPs) by surface‐localized pattern‐recognition receptors (PRRs) activates plant innate immunity, mainly through activation of numerous protein kinases. Appropriate induction of immune responses must be tightly regulated, as many of the kinases involved have an intrinsic high activity and are also regulated by other external and endogenous stimuli. Previous evidences suggest that PAMP‐triggered immunity (PTI) is under constant negative regulation by protein phosphatases but the underlying molecular mechanisms remain unknown. Here, we show that protein Ser/Thr phosphatase type 2A (PP2A) controls the activation of PRR complexes by modulating the phosphostatus of the co‐receptor and positive regulator BAK1. A potential PP2A holoenzyme composed of the subunits A1, C4, and B’η/ζ inhibits immune responses triggered by several PAMPs and anti‐bacterial immunity. PP2A constitutively associates with BAK1 in planta. Impairment in this PP2A‐based regulation leads to increased steady‐state BAK1 phosphorylation, which can poise enhanced immune responses. This work identifies PP2A as an important negative regulator of plant innate immunity that controls BAK1 activation in surface‐localized immune receptor complexes.
The Sainsbury Lab's insight:
Plasma membrane-localized pattern recognition receptors such as FLAGELLIN SENSING2 (FLS2) and EF-TU RECEPTOR (EFR) recognize microbe-associated molecular patterns (MAMPs) to activate the first layer of plant immunity termed pattern-triggered immunity (PTI). A reverse genetics approach with genes responsive to the priming agent β-aminobutyric acid (BABA) revealed IMPAIRED OOMYCETE SUSCEPTIBILITY1 (IOS1) as a critical PTI player. Arabidopsis thaliana ios1 mutants were hypersusceptible to Pseudomonas syringae bacteria. Accordingly, ios1 mutants demonstrated defective PTI responses, notably delayed upregulation of PTI marker genes, lower callose deposition, and mitogen-activated protein kinase activities upon bacterial infection or MAMP treatment. Moreover, Arabidopsis lines overexpressing IOS1 were more resistant to P. syringae and demonstrated a primed PTI response. In vitro pull-down, bimolecular fluorescence complementation, coimmunoprecipitation, and mass spectrometry analyses supported the existence of complexes between the membrane-localized IOS1 and FLS2 and EFR. IOS1 also associated with BRASSINOSTEROID INSENSITIVE1-ASSOCIATED KINASE1 (BAK1) in a ligand-independent manner and positively regulated FLS2/BAK1 complex formation uponMAMP treatment. Finally, ios1 mutants were defective in BABA-induced resistance and priming. This work reveals IOS1 as a regulatory protein of FLS2- and EFR-mediated signaling that primes PTI activation upon bacterial elicitation.
The Sainsbury Lab's insight:
Terrestrial plants rely on stomata, small pores in the leaf surface, for photosynthetic gas exchange and transpiration of water. The stomata, formed by a pair of guard cells, dynamically increase and decrease their volume to control the pore size in response to environmental cues. Stresses can trigger similar or opposing movements: for example, drought induces closure of stomata, whereas many pathogens exploit stomata and cause them to open to facilitate entry into plant tissues. The latter is an active process as stomatal closure is part of the plant's immune response. Stomatal research has contributed much to clarify the signalling pathways of abiotic stress, but guard cell signalling in response to microbes is a relatively new area of research. In this article, we discuss present knowledge of stomatal regulation in response to microbes and highlight common points of convergence, and differences, compared to stomatal regulation by abiotic stresses. We also expand on the mechanisms by which pathogens manipulate these processes to promote disease, for example by delivering effectors to inhibit closure or trigger opening of stomata. The study of pathogen effectors in stomatal manipulation will aid our understanding of guard cell signalling.
The Sainsbury Lab's insight:
Plant PRRs are surface-localized receptor kinases or receptor-like proteins.Plant PRRs recognize a wide range of microbe- or plant-derived molecules.Known plant PAMP/PRR pairs illustrate distinct molecular-recognition mechanisms.PRRs can be used to engineer broad-spectrum disease resistance in crop plants.
Plants are constantly exposed to would-be pathogens in their immediate environment. Yet, despite relying on innate immunity only, plants are resistant to most microbes. They employ pattern-recognition receptors (PRRs) for sensitive and rapid detection of the potential danger caused by microbes and pests. Plant PRRs are either surface-localized receptor kinases (RKs) or receptor-like proteins (RLPs) containing various ligand-binding ectodomains that perceive pathogen-associated molecular patterns (PAMPs) or damage-associated molecular patterns (DAMPs). In this review, I summarize our current knowledge of plant PRRs and their ligands, illustrating the multiple molecular strategies employed by plant PRRs to activate innate immune signaling to survive.
The Sainsbury Lab's insight:
The allohexaploid bread wheat genome consists of three closely related subgenomes (A, B, and D), but a clear understanding of their phylogenetic history has been lacking. We used genome assemblies of bread wheat and five diploid relatives to analyze genome-wide samples of gene trees, as well as to estimate evolutionary relatedness and divergence times. We show that the A and B genomes diverged from a common ancestor ~7 million years ago and that these genomes gave rise to the D genome through homoploid hybrid speciation 1 to 2 million years later. Our findings imply that the present-day bread wheat genome is a product of multiple rounds of hybrid speciation (homoploid and polyploid) and lay the foundation for a new framework for understanding the wheat genome as a multilevel phylogenetic mosaic.

Filamentous pathogens such as the oomycete Phytophthora infestans infect plants by developing specialized structures termed haustoria inside the host cells. Haustoria are thought to enable secretion of effector proteins into the plant cells. Haustorium biogenesis is therefore critical for pathogen accommodation in the host tissue. Haustoria are enveloped by a specialized host-derived membrane, the extrahaustorial membrane (EHM), which is distinct from the plant plasma membrane. The mechanisms underlying the biogenesis of the EHM are unknown. Remarkably, several plasma membrane localised proteins are excluded from the EHM but the remorin REM1.3 accumulates around P. infestans haustoria. Here, we used overexpression, co-localization with reporter proteins, and super-resolution microscopy in cells infected by P. infestans to reveal discrete EHM domains labelled by REM1.3 and P. infestans effector AVRblb2. Moreover, SYT1 synaptotagmin, another previously identified perihaustorial protein, localized to subdomains which are mainly not labelled by REM1.3 and AVRblb2. Functional characterization of REM1.3 revealed that it is a susceptibility factor that promotes infection by P. infestans. This activity, and REM1.3 recruitment to the EHM, require REM1.3 membrane binding domain. Our results implicate REM1.3 membrane micro-domains in plant susceptibility to an oomycete pathogen. Via Kamoun Lab @ TSL

Jean-Michel Ané's curator insight,
May 14, 2014 4:17 PM
I know that it is not a symbiont but... some people will guess why I am scooping this :-) |
The Sainsbury Lab's insight:
Plant nucleotide-binding leucine-rich repeat (NB-LRR) disease resistance (R) proteins recognize specific “avirulent” pathogen effectors and activate immune responses. NB-LRR proteins structurally and functionally resemble mammalian Nod-like receptors (NLRs). How NB-LRR and NLR proteins activate defense is poorly understood. The divergently transcribed Arabidopsis R genes, RPS4 (resistance to Pseudomonas syringae 4) and RRS1 (resistance to Ralstonia solanacearum 1), function together to confer recognition of Pseudomonas AvrRps4 and Ralstonia PopP2. RRS1 is the only known recessive NB-LRR R gene and encodes a WRKY DNA binding domain, prompting suggestions that it acts downstream of RPS4 for transcriptional activation of defense genes. We define here the early RRS1-dependent transcriptional changes upon delivery of PopP2 via Pseudomonas type III secretion. The Arabidopsis slh1 (sensitive to low humidity 1) mutant encodes an RRS1 allele (RRS1SLH1) with a single amino acid (leucine) insertion in the WRKY DNA-binding domain. Its poor growth due to constitutive defense activation is rescued at higher temperature. Transcription profiling data indicate that RRS1SLH1-mediated defense activation overlaps substantially with AvrRps4- and PopP2-regulated responses. To better understand the genetic basis of RPS4/RRS1-dependent immunity, we performed a genetic screen to identify suppressor of slh1 immunity (sushi) mutants. We show that many sushi mutants carry mutations in RPS4, suggesting that RPS4 acts downstream or in a complex with RRS1. Interestingly, several mutations were identified in a domain C-terminal to the RPS4 LRR domain. Using an Agrobacterium-mediated transient assay system, we demonstrate that the P-loop motif of RPS4 but not of RRS1SLH1 is required for RRS1SLH1 function. We also recapitulate the dominant suppression of RRS1SLH1 defense activation by wild type RRS1 and show this suppression requires an intact RRS1 P-loop. These analyses of RRS1SLH1 shed new light on mechanisms by which NB-LRR protein pairs activate defense signaling, or are held inactive in the absence of a pathogen effector.
The Sainsbury Lab's insight:
Importin-αs are essential adapter proteins that recruit cytoplasmic proteins destined for active nuclear import to the nuclear transport machinery. Cargo proteins interact with the importin-α armadillo repeat domain via nuclear localization sequences (NLSs), short amino acids motifs enriched in Lys and Arg residues. Plant genomes typically encode several importin-α paralogs that can have both specific and partially redundant functions. Although some cargos are preferentially imported by a distinct importin-α, it remains unknown how this specificity is generated and to what extent cargos compete for binding to nuclear transport receptors. Here we report that the effector protein HaRxL106 from the oomycete pathogen Hyaloperonospora arabidopsidis co-opts the host cell's nuclear import machinery. We use HaRxL106 as a probe to determine redundant and specific functions of importin-α paralogs from Arabidopsis thaliana. A crystal structure of the importin-α3/MOS6 armadillo repeat domain suggests that five of the six Arabidopsis importin-αs expressed in rosette leaves have an almost identical NLS binding site. Comparison of the importin-α binding affinities of HaRxL106 and other cargos in vitro and in plant cells suggests that relatively small affinity differences in vitro affect the rate of transport complex formation in vivo. Our results suggest that cargo affinity for importin-α, sequence variation at the importin-α NLS binding sites and tissue-specific expression levels of importin-αs determine formation of cargo/importin-α transport complexes in plant cells.
The Sainsbury Lab's insight:
A balance between growth and immunity exists in plants. Recently, the growth-promoting hormones brassinosteroids (BR) have emerged as crucial regulators of the growth-immunity trade-off, although the molecular mechanisms underlying this role remained unclear. New evidence obtained from the model plant Arabidopsis thaliana points at an indirect crosstalk between BR signaling and immunity, mediated by the transcription factors BZR1 and HBI1, which suppress immunity upon BR perception. The core transcriptional cascade formed by BZR1 and HBI1 seems to act as a regulatory hub on which multiple signaling inputs impinge, ensuring effective fine-tuning of the trade-off between growth and immunity in a timely and cost-efficient manner.
The Sainsbury Lab's insight:
Highlights
- Powdery mildew fungus G. orontii virulence effectors and their host-interactors identified - Integrated network map reveals interspecies effector convergence onto shared host proteins - Mutants of convergent effector-targeted host proteins display altered infection phenotypes - Host genes under balancing selection encode indirect targets of pathogen effectors
Summary While conceptual principles governing plant immunity are becoming clear, its systems-level organization and the evolutionary dynamic of the host-pathogen interface are still obscure. We generated a systematic protein-protein interaction network of virulence effectors from the ascomycete pathogen Golovinomyces orontii and Arabidopsis thaliana host proteins. We combined this data set with corresponding data for the eubacterial pathogen Pseudomonas syringae and the oomycete pathogen Hyaloperonospora arabidopsidis. The resulting network identifies host proteins onto which intraspecies and interspecies pathogen effectors converge. Phenotyping of 124 Arabidopsis effector-interactor mutants revealed a correlation between intraspecies and interspecies convergence and several altered immune response phenotypes. Several effectors and the most heavily targeted host protein colocalized in subnuclear foci. Products of adaptively selected Arabidopsis genes are enriched for interactions with effector targets. Our data suggest the existence of a molecular host-pathogen interface that is conserved across Arabidopsis accessions, while evolutionary adaptation occurs in the immediate network neighborhood of effector targets.

PLOS ONE: an inclusive, peer-reviewed, open-access resource from the PUBLIC LIBRARY OF SCIENCE. Reports of well-performed scientific studies from all disciplines freely available to the whole world.
The Sainsbury Lab's insight:
Plants protect themselves against a variety of invading pathogenic organisms via sophisticated defence mechanisms. These responses include deployment of specialized antimicrobial compounds, such as phytoalexins, that rapidly accumulate at pathogen infection sites. However, the extent to which these compounds contribute to species-level resistance and their spectrum of action remain poorly understood. Capsidiol, a defense related phytoalexin, is produced by several solanaceous plants including pepper and tobacco during microbial attack. Interestingly, capsidiol differentially affects growth and germination of the oomycete pathogensPhytophthora infestans and Phytophthora capsici, although the underlying molecular mechanisms remain unknown. In this study we revisited the differential effect of capsidiol on P. infestans and P. capsici, using highly pure capsidiol preparations obtained from yeast engineered to express the capsidiol biosynthetic pathway. Taking advantage of transgenicPhytophthora strains expressing fluorescent markers, we developed a fluorescence-based method to determine the differential effect of capsidiol on Phytophtora growth. Using these assays, we confirm major differences in capsidiol sensitivity between P. infestans and P. capsici and demonstrate that capsidiol alters the growth behaviour of both Phytophthoraspecies. Finally, we report intraspecific variation within P. infestans isolates towards capsidiol tolerance pointing to an arms race between the plant and the pathogens in deployment of defence related phytoalexins.
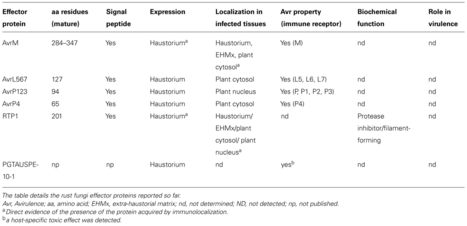
Rust fungi include many species that are devastating crop pathogens. To develop resistant plants, a better understanding of rust virulence factors, or effector proteins, is needed. Thus far, only six rust effector proteins have been described: AvrP123, AvrP4, AvrL567, AvrM, RTP1 and PGTAUSPE-10-1. Although some are well established model proteins used to investigate mechanisms of immune receptor activation (avirulence activities) or entry into plant cells, how they work inside host tissues to promote fungal growth remains unknown. The genome sequences of four rust fungi (two Melampsoraceae and two Pucciniaceae) have been analyzed so far. Genome-wide analyses of these species, as well as transcriptomics performed on a broader range of rust fungi, revealed hundreds of small secreted proteins considered as rust candidate secreted effector proteins (CSEPs). The rust community now needs high-throughput approaches (effectoromics) to accelerate effector discovery/characterization and to better understand how they function in planta. However, this task is challenging due to the non-amenability of rust pathosystems (obligate biotrophs infecting crop plants) to traditional molecular genetic approaches mainly due to difficulties in culturing these species in vitro. The use of heterologous approaches should be promoted in the future.

Oomycetes form a deep lineage of eukaryotic organisms that includes a large number of plant pathogens that threaten natural and managed ecosystems. We undertook a survey to query the community for their ranking of plant pathogenic oomycete species based on scientific and economic importance. In total, we received 263 votes from 62 scientists in 15 countries for a total of 33 species. The Top 10 species and their ranking are: (1) Phytophthora infestans; (2, tied) Hyaloperonospora arabidopsidis; (2, tied) Phytophthora ramorum; (4) Phytophthora sojae; (5) Phytophthora capsici; (6) Plasmopara viticola; (7) Phytophthora cinnamomi; (8, tied) Phytophthora parasitica; (8, tied) Pythium ultimum; and (10) Albugo candida. The article provides an introduction to these 10 taxa and a snapshot of current research. We hope that the list will serve as a benchmark for future trends in oomycete research. See also [link below]:
Top 10 plant-parasitic nematodes in molecular plant pathology
http://onlinelibrary.wiley.com/journal/10.1111/(ISSN)1364-3703/homepage/free_poster.htm Via Kamoun Lab @ TSL
The Sainsbury Lab's insight:
Potato virus Y (PVY, Potyvirus) is the fifth most important plant virus worldwide in terms of economic and scientific impact. It infects members of the family Solanaceae and causes losses in potato, tomato, tobacco, pepper and petunia production. In potato and its wild relatives, two types of resistance genes against PVY have been identified. While Ry genes confer symptomless extreme resistance, Ny genes cause a hypersensitive response visible as local necrosis that may also be able to prevent the virus from spreading under certain environmental conditions. The potato cultivar Sárpo Mira originates from Hungary and is highly resistant to PVY, although the source of this resistance remains unknown. We show that cv. Sárpo Mira reacts with a hypersensitive response leading to necrosis after PVYNTN infection in detached leaf, whole plant and grafting assays. The hypersensitivity to PVYNTN segregated amongst 140 individuals of tetraploid progeny of cvs. Sárpo Mira × Maris Piper in a 1:1 ratio, indicating that it was conferred by a single, dominant gene in simplex. Moreover, we identified five DNA markers linked to this trait and located the underlying locus (Ny-Smira) to the long arm of potato chromosome IX. This position corresponds to the location of the Ry chc and Ny-1 genes for PVY resistance. A simple PCR marker, located 1 cM from the Ny-Smira gene, can be recommended for selection of PVY-resistant progeny of cv. Sárpo Mira.
The Sainsbury Lab's insight:
Plants detect pathogens by sensing microbe-associated molecular patterns (MAMPs) through pattern recognition receptors. Pattern recognition receptor complexes also have roles in cell death control, but the underlying mechanisms are poorly understood. Here, we report isolation of cerk1-4, a novel mutant allele of the Arabidopsis chitin receptor CERK1 with enhanced defense responses.We identified cerk1-4 in a forward genetic screen with barley powdery mildew and consequently characterized it by pathogen assays, mutant crosses and analysis of defense pathways. CERK1 and CERK1-4 proteins were analyzed biochemically.The cerk1-4 mutation causes an amino acid exchange in the CERK1 ectodomain. Mutant plants maintain chitin signaling capacity but exhibit hyper-inducible salicylic acid concentrations and deregulated cell death upon pathogen challenge. In contrast to chitin signaling, the cerk1-4 phenotype does not require kinase activity and is conferred by the N-terminal part of the receptor. CERK1 undergoes ectodomain shedding, a well-known process in animal cell surface proteins. Wild-type plants contain the full-length CERK1 receptor protein as well as a soluble form of the CERK1 ectodomain, whereas cerk1-4 plants lack the N-terminal shedding product.Our work suggests that CERK1 may have a chitin-independent role in cell death control and is the first report of ectodomain shedding in plants.
The Sainsbury Lab's insight:
To identify specific genes determining the initiation and formation of adventitious roots (AR), a microarray-based transcriptome analysis in the stem base of the cuttings of Petunia hybrida (line W115) was conducted. A microarray carrying 24,816 unique, non-redundant annotated sequences was hybridized to probes derived from different stages of AR formation. After exclusion of wound-responsive and root-regulated genes, 1,354 of them were identified which were significantly and specifically induced during various phases of AR formation. Based on a recent physiological model distinguishing three metabolic phases in AR formation, the present paper focuses on the response of genes related to particular metabolic pathways. Key genes involved in primary carbohydrate metabolism such as those mediating apoplastic sucrose unloading were induced at the early sink establishment phase of AR formation. Transcriptome changes also pointed to a possible role of trehalose metabolism and SnRK1 (sucrose non-fermenting 1- related protein kinase) in sugar sensing during this early step of AR formation. Symplastic sucrose unloading and nucleotide biosynthesis were the major processes induced during the later recovery and maintenance phases. Moreover, transcripts involved in peroxisomal beta-oxidation were up-regulated during different phases of AR formation. In addition to metabolic pathways, the analysis revealed the activation of cell division at the two later phases and in particular the induction of G1-specific genes in the maintenance phase. Furthermore, results point towards a specific demand for certain mineral nutrients starting in the recovery phase.
The Sainsbury Lab's insight:
Mass spectrometry (MS) has become the method of choice to identify and quantify proteins, typically by fragmenting peptides and inferring protein identification by reference to sequence databases. Well-established programs have largely solved the problem of identifying peptides in complex mixtures. However, to prevent the search space from becoming prohibitively large most search engines need a list of expected modifications. Therefore, unexpected modifications limit both the identification of proteins and peptide-based quantification. We developed Mass Spectrometry-Peak Shift Analysis (MS-PSA) to rapidly identify related spectra in large datasets without reference to databases or specified modifications. Peptide identifications from established tools, such as MASCOT or SEQUEST, may be propagated onto MS-PSA results. Modification of a peptide alters the mass of the precursor ion and some of the fragmentation ions. MS-PSA identifies characteristic fragmentation masses from MS/MS spectra. Related spectra are identified by pattern matching of unchanged and mass-shifted fragment ions. We illustrate the use of MS-PSA with simple and complex mixtures with both high and low mass accuracy datasets. MS-PSA is not limited to the analysis of peptides but can be used for the identification of related groups of spectra in any set of fragmentation patterns.
The Sainsbury Lab's insight:
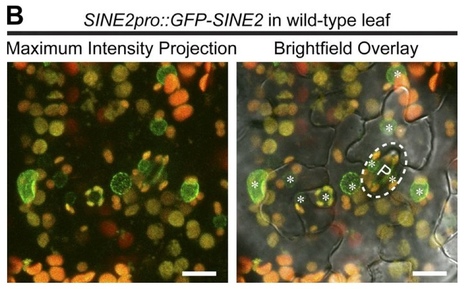
Although a plethora of nuclear envelope (NE) transmembrane proteins (NETs) have been identified in opisthokonts, plant NETs are largely unknown. The only known NET homologues in plants are Sad1/UNC-84 (SUN) proteins, which bind Klarsicht/ANC-1/Syne-1 homology (KASH) proteins. Therefore, de novo identification of plant NETs is necessary. Based on similarities between opisthokont KASH proteins and the only known plant KASH proteins, WPP domain–interacting proteins, we used a computational method to identify the KASH subset of plant NETs. Ten potential plant KASH protein families were identified, and five candidates from four of these families were verified for their NE localization, depending on SUN domain interaction. Of those, Arabidopsis thaliana SINE1 is involved in actin-dependent nuclear positioning in guard cells, whereas its paralogue SINE2 contributes to innate immunity against an oomycete pathogen. This study dramatically expands our knowledge of plant KASH proteins and suggests that plants and opisthokonts have recruited different KASH proteins to perform NE regulatory functions.
|


















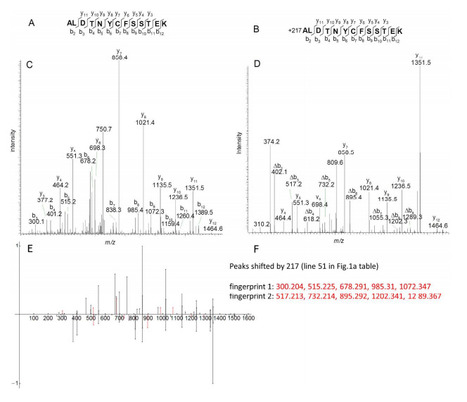





Motivation: Single Nucleotide Polymorphism (SNP) discovery is an important preliminary for understanding genetic variation. With current sequencing methods we can sample genomes comprehensively. SNPs are found by aligning sequence reads against longer assembled references. De Bruijn graphs are efficient data structures that can deal with the vast amount of data from modern technologies. Recent work has shown that the topology of these graphs captures enough information to allow the detection and characterisation of genetic variants, offering an alternative to alignment-based methods. Such methods rely on depth-first walks of the graph to identify closing bifurcations. These methods are conservative or generate many false-positive results, particularly when traversing highly inter-connected (complex) regions of the graph or in regions of very high coverage.
Results: We devised an algorithm that calls SNPs in converted De Bruijn graphs by enumerating 2k + 2 cycles. We evaluated the accuracy of predicted SNPs by comparison with SNP lists from alignment based methods. We tested accuracy of the SNP calling using sequence data from sixteen ecotypes of Arabidopsis thaliana and found that accuracy was high. We found that SNP calling was even across the genome and genomic feature types. Using sequence based attributes of the graph to train a decision tree allowed us to increase accuracy of SNP calls further. Together these results indicate that our algorithm is capable of finding SNPs accurately in complex sub-graphs and potentially comprehensively from whole genome graphs.