 Your new post is loading...

|
Scooped by
?
Today, 11:16 PM
|
In nature, viruses frequently evolve overlapping genes (OLG) in alternate reading frames of the same nucleotide sequence despite the drastically reduced protein sequence space resulting from the sharing of codon nucleotides. Their existence leads one to wonder whether amino acid sequences are sufficiently degenerate with respect to protein folding to broadly allow arbitrary pairs of functional proteins to be overlapped. Here, we investigate this question by engineering synthetic OLGs using state-of-the-art generative models. To evaluate the approach, we first design overlapped sequences targeting two different protein families. We then encode distinct highly ordered de novo protein structures and observe surprisingly high in silico and experimental success rates. This demonstrates that the overlap constraints under the structure of the standard genetic code do not significantly restrict simultaneous accommodation of well defined 3D folds in alternative reading frames. Our work suggests that OLG sequences may be frequently accessible in nature and could be readily exploited to compress and constrain synthetic genetic circuits.
|
|
Scooped by
?
Today, 11:00 PM
|
Archaeal transcription is a hybrid of eukaryotic and bacterial features: a RNAP II-like polymerase transcribes genes organized in circular chromosomes within cells devoid of a nucleus. Consequently, archaeal genomes are depleted in canonical transcription factors (TFs) found in other domains of life. Here we outline the discovery of a cryptic archaea-specific family of ligand-binding TFs, called AmzR (Archaeal Metabolite-sensing Zipper-like Regulators). We identify AmzR through an evolution-based genetic screen and show that it is a negative regulator of methanogenic growth on methylamines in the model archaeon, Methanosarcina acetivorans. AmzR binds its target promoters as an oligomer using paired basic α-helices akin to eukaryotic leucine zippers. AmzR also binds methylamines, which reduces its DNA-binding affinity, and allows it to function as a one-component system commonly found in prokaryotes, while containing a eukaryotic-like DNA-binding motif. The AmzR family of TFs are widespread in archaea and broadens the scope of innovations at the prokaryote-eukaryote interface.
|
|
Scooped by
?
Today, 10:43 PM
|
Increasing drought frequency poses a significant threat to agricultural productivity. A promising strategy to enhance crop resilience against drought is the utilization of root channels left by winter cover crops, which can improve access to subsoil water and nutrients for subsequent cash crops like maize (Zea mays L.). The impact of drought on bacterial communities inhabiting these root channels remains largely unknown. Here, we investigated drought-induced shifts in maize rhizosphere bacterial communities and their functional adaptation in cover crop root channels across three soil types in northern Germany (Luvisol, Podzol, and Phaeozem). Using a multi-omics approach (16S rRNA gene amplicon sequencing, qPCR, and metaproteomics), we identified significant taxonomic and functional responses to drought. A rise in the abundance of K-strategist bacterial communities indicate a shift towards stress-tolerant populations with drought. Under drought stress, the relative abundances of Acidobacteriota, Actinomycetota, Planctomycetota, and Pseudomonadota increased, while Chloroflexota, Methylomirabilota, Ca. Patescibacteria, and Verrucomicrobiota declined. Functional analyses revealed that drought-stressed aerobic taxa among the Pseudomonadota and Verrucomicrobiota upregulated the glyoxylate cycle, potentially enhancing carbon and energy conservation, and increased antioxidant defences (catalase-glutathione peroxidase and methionine cycle-transsulfuration pathway). These drought-mitigating strategies were especially pronounced in root channels formed by Brassicaceae and Poaceae cover crops in the Luvisol and Podzol soils. These findings demonstrate the functional plasticity of rhizosphere bacterial communities in reused root channels in response to drought, highlighting the potential to leverage microbiome-mediated resilience for agricultural practices.
|
|
Scooped by
?
Today, 9:56 PM
|
RRMScorer is a web server designed to predict RNA binding preferences for proteins containing RNA recognition motifs (RRMs), the most prevalent RNA binding domain in eukaryotes. By carefully analysing a dataset of 187 RRM–RNA structural complexes, we calculated residue-level binding scores using a probabilistic model derived from amino acid–nucleotide interaction propensities, which are the basis of RRMScorer. The server accepts protein sequences and optional RNA sequences as input, providing detailed outputs, including bar plots, sequence logos, and downloadable CSV/JSON files, to visualize and interpret RNA binding preferences. RRMScorer is particularly useful for studying the impact of single-point mutations and comparing binding preferences across multiple RRM domains. The web server, freely accessible at https://bio2byte.be/rrmscorer, offers a user-friendly interface and integrates precomputed predictions for over 1400 RRM-containing proteins. With its ability to provide residue-level insights and accurate predictions, RRMScorer serves as a valuable tool for researchers exploring the functional landscape of RRM–RNA interactions.
|
|
Scooped by
?
Today, 9:49 PM
|
Finding appropriate software and parameter settings to process shotgun metagenome data is essential for meaningful metagenomic analyses. To enable objective and comprehensive benchmarking of metagenomic software, the community-led initiative for the Critical Assessment of Metagenome Interpretation (CAMI) promotes standards and best practices. Since 2015, CAMI has provided comprehensive datasets, benchmarking guidelines, and challenges. However, benchmarking had to be conducted offline, requiring substantial time and technical expertise and leading to gaps in results between challenges. We introduce the CAMI Benchmarking Portal—a central repository of CAMI resources and web server for the evaluation and ranking of metagenome assembly, binning, and taxonomic profiling software. The portal simplifies evaluation, enabling users to easily compare their results with previous and other users’ submissions through a variety of metrics and visualizations. As a demonstration, we benchmark software performance on the marine dataset of the CAMI II challenge. The portal currently hosts 28 675 results and is freely available at https://cami-challenge.org/.
|
|
Scooped by
?
Today, 9:44 PM
|
The UniProt REST API is a freely available, open-access resource that powers the UniProt.org website and gives users flexible programmatic interaction with protein knowledge data. It provides access to UniProtKB, UniRef, UniParc, Proteomes, GeneCentric, ARBA, UniRule, and the ID Mapping tool, along with supporting data and controlled vocabularies. Users can access the API with their favorite programming language and generate example code snippets to access the UniProt databases using the API documentation page (https://www.uniprot.org/api-documentation) in various languages. API results can be returned and downloaded in various formats. With an average of 303 million requests per month over the last year, the API enables structured search queries using logical operators and parentheses, allows users to specify fields of interest within results, and customize downloads for direct integration into workflows. The API is a powerful tool that empowers users to fully utilize UniProt data across multiple datasets, enabling download, analysis, and extraction of valuable research insights. This website is free and open to all users, and there is no login requirement.
|
|
Scooped by
?
Today, 5:46 PM
|
Identifying beneficial or functional mutations for a specific species is a task always relies on the labor-intensive construction of a library containing thousands of single-gene knockout or interference strains. Here, we systematically demonstrated that the task could be done by constructing a genome-scale library, designed by AutoGSL, containing a limited number of large fragment deletion strains. The loss-of- and gain-of-function phenotypes could be efficiently screened out by our chromosome-segment scanning, and the specific gene corresponding to an observed phenotype could be quickly identified and visualized by our computer-aided gene-annotation web tool. Additionally, a Clusters of Orthologous Gene Transformer learned representations were transferred to predict growth phenotypes of the genome-scale library under varying conditions. We further utilized our chromosome segment scanning for gain- or loss-of-function screening (CHASING) strategy to obtain acetoin- and lycopene-overproducing hosts. Our work highlighted the significance of CHASING in functional genomics investigation, robust chassis engineering, and chemical overproduction.
|
|
Scooped by
?
Today, 5:33 PM
|
Alternative food products are needed to address the most pressing challenges faced by the food industry: growing global food demand, health concerns, animal welfare, food security and environmental sustainability. Future foods are defined as foods with scalability and sustainability potential owing to rapidly advancing technological developments in their production systems. Key areas of study for future foods include cellular agriculture and plant-based systems, which include biomaterials as key ingredients or as structural components to impart texture, support cell growth and metabolism, and provide nutrients and organoleptic factors to food products. This Review discusses current requirements, options and processing approaches for biomaterials with utility in future foods. We focus on two main approaches: cellular agriculture wherein the cells are the key component for food (with the biomaterials utilized to support the cells via adherence and/or for texture) and plant-based foods wherein acellular plant-derived biomaterials are the food components. In both cases, the same fundamental challenges apply for the biomaterials: achieving utility at scale and low cost while meeting food safety requirements. Other considerations for biomaterials for future foods are also addressed, including sustainability, modelling, consumer acceptance, nutrition, regulatory status and safety considerations to highlight the path ahead. This emerging field of biomaterials for future foods offers a new generation of biomaterial systems that can positively impact human health, environmental sustainability and animal welfare. Although scaling these biomaterial sources cost-effectively presents a major challenge, substantial progress is being made, and opportunities to establish supply chains are already underway. Biomaterials have a crucial role in the development of future foods, particularly in cellular agriculture and plant-based systems. This Review addresses the current status and future requirements of biomaterials for future foods, addressing key aspects such as structure, nutrition, safety, sensory attributes, sustainability and consumer preferences.
|
|
Scooped by
?
Today, 1:16 AM
|
A key unresolved question in microbial ecology is how the extraordinary diversity of microbiomes emerges from the interactions among their many functionally distinct populations. This process is driven in part by the cross-feeding networks that help to structure these systems, in which consumers use resources to fuel their metabolism, creating by-products which can be used by others in the community. Understanding the effects of cross-feeding presents a major challenge, as it creates complex interdependencies between populations which can be hard to untangle. We address this problem using the tools of network science to develop a structural microbial community model. Using methods from percolation theory, we identify feasible community states for cross-feeding network structures in which the needs of consumers are met by metabolite production across the community. We identify tipping points at which small changes in structure can cause the catastrophic collapse of cross-feeding networks and abrupt declines in microbial community diversity. Our results are an example of a well-defined tipping point in a complex ecological system and provide insight into the fundamental processes shaping microbiomes and their robustness. We further demonstrate this by considering how network attacks affect community diversity and apply our results to show how the apparent difficulty in culturing the microbial diversity emerges as an inherent property of their cross-feeding networks.
|
|
Scooped by
?
Today, 1:05 AM
|
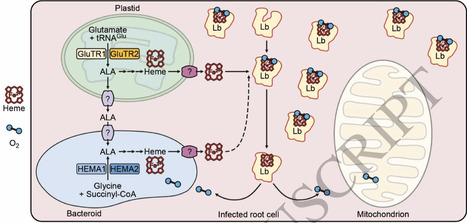
Heme is biosynthesized in legume root nodules to meet the demand for leghemoglobins (Lbs) and other heme-binding proteins. However, the main source of nodule heme remains unknown. Both the plant host and rhizobia possess a complete heme biosynthetic pathway, differing slightly in the production of 5-aminolevulinic acid (ALA), a key regulatory step catalyzed by glutamyl-tRNA reductase (GluTR) in the plant and by HemA in the rhizobia. Transcriptomic analysis revealed that many plant heme biosynthetic genes, including GluTR2 but not GluTR1, are upregulated in nodules compared to roots, whereas expression of related rhizobial genes, including both HemA1 and HemA2, is generally inhibited under symbiotic conditions compared to free-living conditions. Knockout of Lotus japonicus GluTR2, but not of HemA1 and HemA2, led to a significant decrease (∼50%) in nodule heme content. The stable heterozygous mutant of GluTR1 or transient knockdown of GluTR1 exhibited a ∼20% reduction in nodule heme content. Overexpression of Fluorescent in blue light (FLU), a feedback inhibitor of GluTR activity, caused a much greater reduction in nodule heme content (∼75%) and an increased level of apo-Lb and, in combination with the hemA1 hemA2 mutant, a drastic inhibition of nitrogenase activity (>90%). This study provides genetic evidence supporting a major role of plant GluTRs in coordinating heme biosynthesis between the two symbionts by supplying heme to assemble with cytoplasmic apo-Lbs and by providing ALA for heme synthesis in the bacteroids.
|
|
Scooped by
?
Today, 12:51 AM
|
Plant-associated microorganisms interact with each other and host plants via intricate chemical signals, offering multiple benefits, including plant nutrition. We report such a mechanism through which the rhizosphere microbiome improves plant growth under sulfur (S) deficiency. Disruption of plant S homeostasis resulted in a coordinated shift in composition and S-metabolism of the rhizosphere microbiome. Using this insight, we developed an 18-membered synthetic rhizosphere bacterial community (SynCom) that rescued growth of Arabidopsis and a leafy Brassicaceae vegetable under S-deficiency. This beneficial trait is taxonomically widespread among SynCom members, with bacterial pairs predominantly providing synergistic benefits to host plants. Notably, stronger competitive interactions among SynCom members conferred greater fitness benefits to the host, suggesting a trans-kingdom fitness tradeoff. Finally, guided chemical screening, deletion knockout mutant, and targeted metabolomics identified and validated microbially released glutathione (GSH) as the necessary bioactive signal that orchestrates plant-microbe (trans-kingdom) fitness tradeoff and improves plant growth under sulfur limitation.
|
|
Scooped by
?
Today, 12:37 AM
|
Phages are typically classified as temperate, integrating into host genomes, or lytic, replicating and killing bacteria. Lytic phages are not expected in bacterial genome sequences, yet our analysis of 3.6 million bacterial assemblies from 1,226 species revealed >100,000 complete lytic phage genomes, which we term Bacterial Assembly-associated Phage Sequences (BAPS). This represents a ~five-fold increase in the number of phages with known hosts and fundamentally reshapes our understanding of phage biology. Identifying BAPS has enabled the discovery of entirely novel phage clusters, including clusters distantly related to Salmonella Goslarviruses in E. coli, and Shigella, while significantly expanding known genera such as Seoulvirus (from 16 to >300 members). Notably, close relatives of therapeutic lytic phages were also detected, suggesting clinical isolate sequencing unknowingly archives viable phage candidates. The discovery of complete, lytic phage genomes within bacterial assemblies challenges long-standing assumptions and reveals a vast, untapped reservoir of phages.
|
|
Scooped by
?
Today, 12:31 AM
|
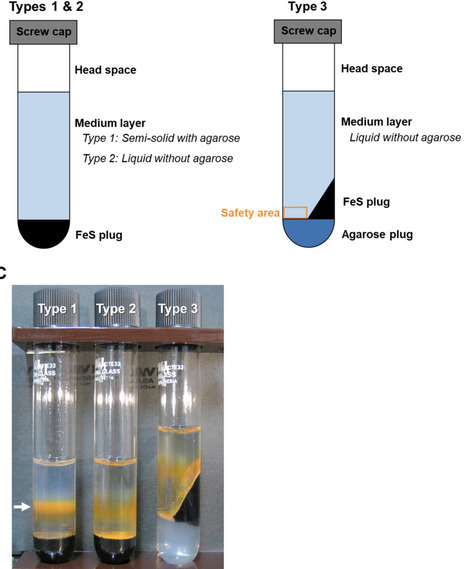
Chemolithotrophic neutrophilic iron (Fe)-oxidizing bacteria, which mainly belong to the family Gallionellaceae, universally prevail in terrestrial environments changing iron cycling. However, they are typically recognized as difficult-to-culture microbes. Despite efforts, there are few Fe(II)-oxidizing lithotroph isolates; hence, their physiological and ecological knowledge remains limited. This limitation is largely owing to difficulties in their cultivation, and we hypothesize that the difficulty exists because substrate and mineral concentrations in the cultivation medium are not tuned to a specific environmental condition under which these organisms live. To address this hypothesis, this study proposes a novel custom-made medium approach for chemolithotrophic Fe(II)-oxidizing bacteria; a method that manipulates medium components through diligent analysis of field environment. A new custom-made medium simulating energy substrates and nutrients under the field condition was prepared by modifying both chemical composition and physical setup in the glass-tube medium. In particular, the modification of the physical setup in the tube had a significant effect on adjusting dissolved Fe(II) and O2 concentrations to the field environment. Using the medium, Gallionellaceae members were successfully enriched and a new Gallionellaceae species was isolated from a natural hot spring site. Compared with conventional medium, the custom-made medium has significantly higher ability in enriching Gallionellaceae members.
|
|
|
Scooped by
?
Today, 11:13 PM
|
Bacillus methanolicus represents a thermophilic methylotroph whose methanol utilization depends on plasmid-encoded genes. It serves as a unique model for deciphering plasmid-dependent methylotrophy and an ideal chassis for low-carbon biomanufacturing using CO2-derived C1 substrates. Despite its evolutionary uniqueness and industrial potential, the lack of synthetic biology tools has hindered both mechanistic understanding and strain engineering. Here, we present a comprehensive synthetic biology platform comprising a high-efficiency electroporation protocol, a CRISPR method enabling robust and multiplex genome editing, diverse neutral loci for gene integration and overexpression, and a cloud-based genome-scale metabolic model iBM822 for user-friendly biodesign. Leveraging this toolkit, we systematically dissected plasmid-dependent methylotrophy, host restriction-modification systems, and functional significance of the chromosomal methylotrophic genes through targeted deletion. To address plasmid loss-induced strain degeneration, we integrated the large endogenous plasmid pBM19 into the chromosome for stable and intact methylotrophic growth. Finally, by integrating metabolic modeling with CRISPR editing, we engineered L-arginine feedback regulation to achieve the first L-arginine biosynthesis from methanol. This study establishes a synthetic biology framework for B. methanolicus, promoting mechanistic exploration of methylotrophy and low-carbon biomanufacturing.
|
|
Scooped by
?
Today, 10:47 PM
|
Formate is a single-carbon compound that is challenging to assimilate, including when assimilation involves a CO2 intermediate that can diffuse from the cell. Mutations that overcome such challenges can be identified through adaptive laboratory evolution. We evolved the anoxygenic phototrophic bacterium Rhodopseudomonas palustris to use formate as the sole carbon source. Through gene deletions, we determined that formate is assimilated via oxidation to CO2 by formate dehydrogenase followed by CO2 fixation by the Calvin cycle. However, this pathway had no clear link to three genes that were commonly mutated in evolved isolates: (i) ribB, a flavin synthesis enzyme, (ii) ppsR2, a repressor of light-harvesting genes, and (iii) RPA0893, a regulator of unknown function. A RibB mutation was necessary and sufficient for formate assimilation and improved formate oxidation. PpsR2 mutations occurred early, facilitated formate assimilation, and caused elevated pigmentation. Pigment production generates CO2 and alkaline conditions that, along with intracellular chromatophore membranes, could represent a rudimentary but important CO2-concentrating mechanism. RPA0983 mutations emerged late and facilitated formate assimilation. RPA0983 proximity to a CO2-liberating pigment synthesis gene suggests a similar effect as PpsR2 mutations. Our findings reveal unintuitive pathway intersections that could have broad implications for formate and CO2-utilizing organisms.
|
|
Scooped by
?
Today, 9:58 PM
|
Industrial wastewater, petroleum pollution and plastic contamination are significant threats to global marine biosecurity because of their toxic, mutagenic and persistent nature. The use of microorganisms in bioremediation has been constrained by the complexity of organic pollutants and limited tolerance to saline stress. In this study, we used synthetic biology to engineer Vibrio natriegens into a strain capable of bioremediating complex organic pollutants in saline wastewater and soils. The competence master regulator gene tfoX was inserted into chromosome 1 of the V. natriegens strain Vmax and overexpressed to enhance DNA uptake and integration. Degradation gene clusters were chemically synthesized and assembled in yeast. We developed a genome engineering method (iterative natural transformation based on Vmax with amplified tfoX effect) to transfer five gene clusters (43 kb total) into Vmax. The engineered strain has the ability to bioremediate five organic pollutants (biphenyl, phenol, naphthalene, dibenzofuran and toluene) covering a broad substrate range, from monocyclic to multicyclic compounds, in industrial wastewater samples from a chlor–alkali plant and a petroleum refinery. A synthetic biology approach was used to engineer Vibrio natriegens into a strain capable of bioremediating complex organic pollutants in saline wastewater and soils, thereby addressing notable threats to marine biosecurity.
|
|
Scooped by
?
Today, 9:53 PM
|
Accurate protein structure alignment is essential for understanding structural and functional relationships. Here, we introduce GTalign-web, a web-based implementation of GTalign, a spatial index-driven protein structure alignment tool, designed for accessibility and high-performance structural searches. Benchmarked against the DALI and Foldseek servers, GTalign-web demonstrates superior accuracy while maintaining rapid search times. Its utility is further highlighted in annotating uncharacterized proteins through searches against UniRef30. GTalign-web provides a useful resource for protein structure analysis and functional annotation and is available at https://bioinformatics.lt/comer/gtalign.
|
|
Scooped by
?
Today, 9:46 PM
|
With the growing number of AI-predicted protein structures, automated methods of broad-scale analysis are required to parse this volume of data. The application of mathematically defined topologies to protein science enables such analysis. Building on the foundation of lasso peptides, complex lasso motifs are their macroscopic analogs in proteins, promising novel discoveries in drug design and the biopolymer industry. Here we present AlphaLasso, a web server designed to find and analyze lasso-type topologies in protein structures. It finds cysteine, amide, ester, and thioester or user-specified closing bridges. The modern visualization interface provides extensive capabilities to study lasso motifs, such as structure smoothing, creating topology maps, searching for similar proteins, in-depth model evaluation, and metadata annotation. This rich feature set makes AlphaLasso a powerful tool useful in biology, biophysics, chemistry, and mathematics. To enable large-scale analysis, we have precomputed the lasso topologies of high-quality models from the AlphaFold Database, finding >14 million proteins with lasso motifs closed by cysteine bridges, 2.2 million of which are complex lassos. Lasso motifs classified by complexity are available to users via an interactive website, supporting comparison with user-submitted structures. AlphaLasso is available at https://alphalasso.cent.uw.edu.pl/.
|
|
Scooped by
?
Today, 9:31 PM
|
As members of the α-proteobacteria group, Caulobacter crescentus and its relatives are wildly studied for their unique asymmetric life cycle and versatile applications in industry, agriculture, and biomedicine. However, genetic manipulation in these bacteria remains challenging, typically requiring time-consuming and labor-intensive procedures. Here, we report a practical CRISPR-SpCas9M-reporting system that overcomes the limitations of SpCas9 expression and CRISPR escape, enabling efficient, markerless, and rapid genome editing in C. crescentus. Two genes encoding for a pair of scaffold proteins were knocked out individually or iteratively, demonstrating their direct involvements in cellular signaling asymmetry. Key components, including the Cas protein, Cas inducer, sgRNA, homologous arms, and reporter, were systematically analyzed and optimized in the system, finally achieving the apparent editing efficiency up to 80% in C. crescentus. Furthermore, we applied the CRISPR-SpCas9M-reporting system to two C. crescentus relatives, Agrobacterium fabrum and Sinorhizobium meliloti, establishing it as an efficient and general editing strategy. We anticipate that this system could be applied to other CRISPR-Cas-recalcitrant organisms, accelerating both basic and applied research in α-proteobacteria.
|
|
Scooped by
?
Today, 5:37 PM
|
Studies of membrane protein folding have progressed from simple systems such as bacteriorhodopsin to complex structures such as ATP-binding cassette transporters and voltage-gated ion channels. Advances in techniques such as single-molecule force spectroscopy and in vivo force profiling now allow for the detailed examination of membrane protein folding pathways at amino acid resolutions. These proteins navigate rugged energy landscapes partly shaped by the absence of hydrophobic collapse and the viscous nature of the lipid bilayer, imposing biophysical limitations on folding speeds. Furthermore, many transmembrane (TM) helices display reduced hydrophobicity to support functional requirements, simultaneously increasing the energy barriers for membrane insertion, a manifestation of the evolutionary trade-off between functionality and foldability. These less hydrophobic TM helices typically insert and fold as helical hairpins, following the protein synthesis direction from the N terminus to the C terminus, with assistance from endoplasmic reticulum (ER) chaperones like the Sec61 translocon and the ER membrane protein complex. The folding pathways of multidomain membrane proteins are defined by allosteric networks that extend across various domains, where mutations and folding correctors affect seemingly distant domains. A common evolutionary strategy is likely to be domain specialization, where N-terminal domains enhance foldability and C-terminal domains enhance functionality. Thus, despite inherent biophysical constraints, evolution has finely tuned membrane protein sequences to optimize foldability, stability, and functionality.
|
|
Scooped by
?
Today, 5:25 PM
|
Nitrous oxide (N2O) is the third most important greenhouse gas and originates primarily from natural and engineered microbiomes. Effective emission mitigations are currently hindered by the largely unresolved ecophysiological controls of coexisting N2O-converting metabolisms in complex communities. To address this, we used biological wastewater treatment as a model ecosystem and combined long-term metagenome-resolved metaproteomics with ex situ kinetic and full-scale operational characterization over nearly 2 years. By leveraging the evidence independently obtained at multiple ecophysiological levels, from individual genetic potential to actual metabolism and emergent community phenotype, the cascade of environmental and operational triggers driving seasonal N2O emissions has ultimately been resolved. We identified nitrifier denitrification as the dominant N2O-producing pathway and dissolved O2 as the prime operational parameter, paving the way to the design and fostering of robust emission control strategies. This work exemplifies the untapped potential of multi-meta-omics in the mechanistic understanding and ecological engineering of microbiomes towards reducing anthropogenic impacts and advancing sustainable biotechnological developments. Nitrous oxide is one of the main greenhouse gases emitted during biological wastewater treatment. This long-term multi-meta-omics analysis of a wastewater treatment plant identifies the main causes of nitrous oxide emissions and actionable mitigation strategies.
|
|
Scooped by
?
Today, 1:08 AM
|
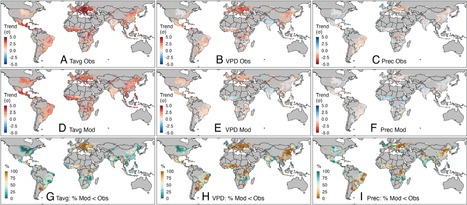
Efforts to anticipate and adapt to future climate can benefit from historical experiences. We examine agroclimatic conditions over the past 50 y for five major crops around the world. Most regions experienced rapid warming relative to interannual variability, with 45% of summer and 32% of winter crop area warming by more than two SD (σ). Vapor pressure deficit (VPD), a key driver of plant water stress, also increased in most temperate regions but not in the tropics. Precipitation trends, while important in some locations, were generally below 1σ. Historical climate model simulations show that observed changes in crops’ climate would have been well predicted by models run with historical forcings, with two main surprises: i) models substantially overestimate the amount of warming and drying experienced by summer crops in North America, and ii) models underestimate the increase in VPD in most temperate cropping regions. Linking agroclimatic data to crop productivity, we estimate that climate trends have caused current global yields of wheat, maize, and barley to be 10, 4, and 13% lower than they would have otherwise been. These losses likely exceeded the benefits of CO2 increases over the same period, whereas CO2 benefits likely exceeded climate-related losses for soybean and rice. Aggregate global yield losses are very similar to what models would have predicted, with the two biases above largely offsetting each other. Climate model biases in reproducing VPD trends may partially explain the ineffectiveness of some adaptations predicted by modeling studies, such as farmer shifts to longer maturing varieties.
|
|
Scooped by
?
Today, 12:56 AM
|
Polyhydroxyalkanoates are biopolyesters synthesized and stored in intracellular granules by diverse prokaryotes. Despite intense research efforts and prior evidence of a rather widespread phylogenetic occurrence of the related genetic machinery, reports on extreme thermophilic and hyperthermophilic polyhydroxyalkanoates producers remain scarce. However, thermophilic cell factories for bioplastic production would serve as an excellent example of Next-Generation Industrial Biotechnology. In this study, we aim to address this research gap by establishing a bioinformatics pipeline to mine genomes of extremely and moderately thermophilic microorganisms for signatures of potential polyhydroxyalkanoate PHB production. Based on a collection of verified protein sequences of polyhydroxyalkanoate polymerase PhaC, the key biosynthetic enzyme, carefully curated sets of thermophilic bacterial and archaeal genomes were screened. This revealed that although PhaC-encoding genes are prevalent in diverse moderately thermophilic bacteria, they are absent in the considered extreme thermophilic bacteria. In contrast, a few limited examples of extreme thermophilic archaea were found to encode putative phaC genes embedded within a typical polyhydroxyalkanoate synthesis operon in their genomes, namely within the genera Ferroglobus, Geoglobus and Archaeoglobus, while no hits were found in extreme thermophilic bacteria. The latter included Thermus thermophilus, which was previously reported as a polyhydroxyalkanoates producer. This was refuted in our bioinformatics analysis and moreover, the predicted absence of polyhydroxyalkanoates synthesis in T. thermophilus was experimentally confirmed by employing various extraction and analytical methods. Based on the findings in this study, we conclude that polyhydroxyalkanoate production is very scarce in extreme thermophiles and hyperthermophiles, for reasons that remain to be elucidated.
|
|
Scooped by
?
Today, 12:44 AM
|
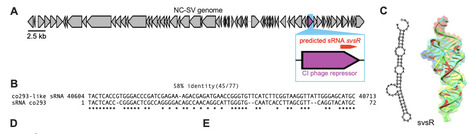
Prophages are prevalent features of bacterial genomes that can reduce susceptibility to lytic phage infection, yet the mechanisms involved are often elusive. Here, we identify a small RNA (svsR) encoded by the lambdoid prophage NC-SV in adherent-invasive Escherichia coli (AIEC) strain NC101 that confers resistance to lytic coliphages. Comparative genomic analyses revealed that NC-SV-like prophages and svsR homologs are conserved across diverse Enterobacteriaceae. Transcriptional analyses reveal that svsR represses maltodextrin transport genes, including lamB, which encodes the outer membrane maltoporin LamB--a known receptor for multiple phages. Nutrient supplementation experiments show that maltodextrin enhances phage adsorption, while glucose suppresses it, consistent with established effects of these sugars on lamB expression. In vivo, we compared wild-type NC101 and a prophage-deletion strain (NC101deltaNC-SV) in mice to assess the impact of NC-SV on lytic phage susceptibility. Although intestinal E. coli densities remained stable across groups, animals colonized with NC101 exhibited markedly reduced phage burdens in both the intestinal lumen and mucosa compared to mice colonized with NC101deltaNC-SV. This reduced phage pressure was associated with increased dissemination of NC101 to extraintestinal tissues, including the spleen and liver. Together, these findings highlight a nutrient-responsive, prophage-encoded mechanism that protects AIEC from phage predation and may promote bacterial persistence and dissemination in the inflamed gut.
|
|
Scooped by
?
Today, 12:35 AM
|
Over the past two decades, metagenomics has greatly expanded our understanding of microbial phylogenetic and metabolic diversity. However, most microbial taxa remain uncultured, hindering research and biotechnological applications. Isolating environmental anaerobes using traditional methods is particularly cumbersome and low throughput. Here, we present a novel, high-throughput approach for the cultivation and isolation of anaerobes, which involves trapping and growing single microbes within selectively permeable hydrogel capsules followed by fluorescence-activated cell sorting to distribute compartmentalized isolates into liquid medium for further growth. We show that diverse anaerobes can grow within capsules and that slower-growing ones (e.g. methanogens) can be enriched with this platform. We also applied this approach to isolate anaerobes from soil, including strains of the sulfate-reducing bacteria Desulfovibrio desulfuricans and Nitratidesulfovibrio vulgaris. Overall, this work introduces a robust, high-throughput alternative to traditional techniques for isolating environmental anaerobes and expands the emerging set of microfluidics-based tools for the cultivation of novel taxa.
|






























crossfeed