Your new post is loading...

|
Scooped by
?
April 25, 2021 4:23 PM
|
Rice leaf blight is caused by the bacterium Xanthomonas oryzae pv. oryzae (Xoo). The upregulated by transcription activator-like 1 (UPT) effector box in the promoter region of the rice Xa13 gene plays a key role in Xoo pathogenicity. Mutation of a key bacterial protein-binding site in the UPT box of Xa13 to abolish PXO99-induced Xa13 expression is a way to improve rice resistance to bacteria. Highly efficient generation and selection of transgene-free edited plants are helpful to shorten and simplify the gene editing-based breeding process. Selective elimination of transgenic pollen of T0 plants can enrich the proportion of T1 transgene-free offspring, and expression of a color marker gene in seeds makes the selection of T2 plants very convenient and efficient. In this study, a genome editing and multiplexed selection system was used to generate bacterial leaf blight-resistant and transgene-free rice plants. We introduced site-specific mutations into the UPT box using CRISPR/Cas12a technology to hamper with transcription-activator-like effector (TAL) protein binding and gene activation and generated genome-edited rice with improved bacterial blight resistance. Transgenic pollen of T0 plants was eliminated by pollen-specific expression of the α-amylase gene Zmaa1, and the proportion of transgene-free plants increased from 25 to 50% among single T-DNA insertion events in the T1 generation. Transgenic seeds were visually identified and discarded by specific aleuronic expression of DsRed, which reduced the cost by 50% and led to up to 98.64% accuracy for the selection of transgene-free edited plants.
|
|
Scooped by
?
April 25, 2021 3:06 PM
|
- Cable bacteria are sulfide‐oxidizing, filamentous bacteria which reduce toxic sulfide levels, suppress methane emissions, and drive nutrient and carbon cycling in sediments. Recently, cable bacteria have been found associated with roots of aquatic plants and rice (Oryza sativa). However, the extent to which cable bacteria are associated with aquatic plants in nature remains unexplored.
- Using newly generated and public 16S rRNA gene sequence datasets combined with fluorescence in situ hybridization, we investigated the distribution of cable bacteria around the roots of aquatic plants, encompassing seagrass (including seagrass seedlings), rice, freshwater and saltmarsh plants.
- Diverse cable bacteria were found associated with roots of 16 out of 28 plant species and at 36 out of 55 investigated sites, across four continents. Plant associated cable bacteria were confirmed across a variety of ecosystems, including marine coastal environments, estuaries, freshwater streams, isolated pristine lakes and intensive agricultural systems. This pattern indicates that this plant‐microbe relationship is globally widespread and neither obligate nor species‐specific.
- The occurrence of cable bacteria in plant rhizospheres may be of general importance to vegetation vitality, primary productivity, coastal restoration practices and greenhouse gas balance of rice fields and wetlands.
|
|
Scooped by
?
April 25, 2021 2:10 PM
|
The RNA chaperone Hfq, acting as a hexamer, is a known mediator of post-transcriptional regulation, expediting basepairing between small RNAs (sRNAs) and their target mRNAs. However, the intricate details associated with Hfq-RNA biogenesis are still unclear. Previously, we reported that the stringent response regulator, RelA, is a functional partner of Hfq that facilitates Hfq-mediated sRNA–mRNA regulation in vivo and induces Hfq hexamerization in vitro. Here we show that RelA-mediated Hfq hexamerization requires an initial binding of RNA, preferably sRNA to Hfq monomers. By interacting with a Shine–Dalgarno-like sequence (SDL, GGAG) in the sRNA, RelA stabilizes the initially unstable complex of RNA bound-Hfq monomer, enabling the attachment of more Hfq subunits to form a functional hexamer. Overall, our study showing that RNA binding to Hfq monomers is at the heart of RelA-mediated Hfq hexamerization, challenges the previous concept that only Hfq hexamers can bind RNA.
|
|
Scooped by
?
April 23, 2021 2:19 AM
|
When plants face an environmental stress such as water deficit, soil salinity, high temperature, or shade, good communication between above- and belowground organs is necessary to coordinate growth and development. Various signals including hormones, peptides, proteins, hydraulic signals, and metabolites are transported mostly through the vasculature to distant tissues. How shoots and roots synchronize their response to stress using mobile signals is an emerging field of research. We summarize recent advances on mobile signals regulating shoot stomatal movement and root development in response to highly localized environmental cues. In addition, we highlight how the vascular system is not only a conduit but is also flexible in its development in response to abiotic stress.
|
|
Scooped by
?
April 23, 2021 12:44 AM
|
Ribosome profiling spectra bear rich information on translation control and dynamics. Yet, due to technical biases in library generation, extracting quantitative measures of discrete translation events has remained elusive. Using maximum likelihood statistics and data set from Escherichia coli we develop a robust method for neutralizing technical biases (e.g. base specific RNase preferences in ribosome-protected mRNA fragments (RPF) generation), which allows for correct estimation of translation times at single codon resolution. Furthermore, we validated the method with available datasets from E. coli treated with antibiotic to inhibit isoleucyl-tRNA synthetase, and two datasets from Saccharomyces cerevisiae treated with two RNases with distinct cleavage signatures. We demonstrate that our approach accounts for RNase cleavage preferences and provides bias-corrected translation times estimates. Our approach provides a solution to the long-standing problem of extracting reliable information about peptide elongation times from highly noisy and technically biased ribosome profiling spectra.
|
|
Scooped by
?
April 22, 2021 10:00 PM
|
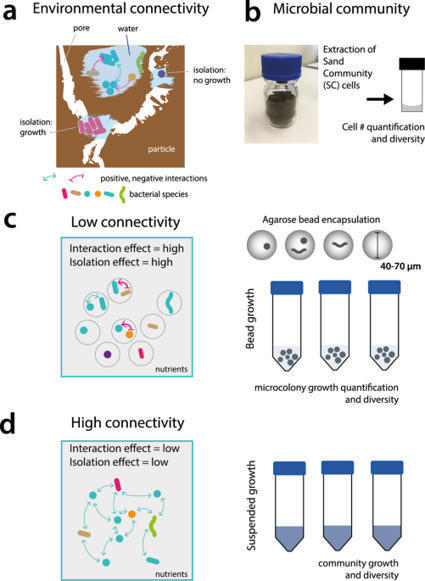
Interspecific interactions are thought to govern the stability and functioning of microbial communities, but the influence of the spatial environment and its structural connectivity on the potential of such interactions to unfold remain largely unknown. Here we studied the effects on community growth and microbial diversity as a function of environmental connectivity, where we define environmental connectivity as the degree of habitat fragmentation preventing microbial cells from living together. We quantitatively compared growth of a naturally-derived high microbial diversity community from soil in a completely mixed liquid suspension (high connectivity) to growth in a massively fragmented and poorly connected environment (low connectivity). The low connectivity environment consisted of homogenously-sized miniature agarose beads containing random single or paired founder cells. We found that overall community growth was the same in both environments, but the low connectivity environment dramatically reduced global community-level diversity compared to the high connectivity environment. Experimental observations were supported by community growth modeling. The model predicts a loss of diversity in the low connectivity environment as a result of negative interspecific interactions becoming more dominant at small founder species numbers. Counterintuitively for the low connectivity environment, growth of isolated single genotypes was less productive than that of random founder genotype cell pairs, suggesting that the community as a whole profited from emerging positive interspecific interactions. Our work demonstrates the importance of environmental connectivity for growth of natural soil microbial communities, which aids future efforts to intervene in or restore community composition to achieve engineering and biotechnological objectives.
|
|
Scooped by
?
April 22, 2021 9:07 AM
|
-
Sustainable food systems will require profound changes in people’s consumption patterns and lifestyles, which is true regardless of the farming methods used and does not change the fact that organic farming often requires more land than conventional farming for the same quantity of food output. -
Some features of organic farming in the EU contribute to the Sustainable Development Goals (SDGs); other features may jeopardize the achievement of SDGs 2, 13, and 15. The negative indirect effects of additional land-use change may outweigh the positive direct effects on global climate and biodiversity, so that a large-scale switch to organic farming in the EU could possibly turn out to be a disservice to global sustainability. -
Achieving the SDGs would benefit from the inclusion of biotech innovations in organic farming. -
The implementation of required changes in the EU law is unlikely under current political realities but is nevertheless recommended from a scientific perspective.
|
|
Scooped by
?
April 22, 2021 2:14 AM
|
Metagenomics has yielded massive amounts of sequencing data offering a glimpse into the biosynthetic potential of the uncultivated microbial majority. While genome-resolved information about microbial communities from nearly every environment on earth is now available, the ability to accurately predict biocatalytic functions directly from sequencing data remains challenging. Compared to primary metabolic pathways, enzymes involved in secondary metabolism often catalyze specialized reactions with diverse substrates, making these pathways rich resources for the discovery of new enzymology. To date, functional insights gained from studies on environmental DNA (eDNA) have largely relied on PCR- or activity-based screening of eDNA fragments cloned in fosmid or cosmid libraries. As an alternative, shotgun metagenomics holds underexplored potential for the discovery of new enzymes directly from eDNA by avoiding common biases introduced through PCR- or activity-guided functional metagenomics workflows. However, inferring new enzyme functions directly from eDNA is similar to searching for a ‘needle in a haystack’ without direct links between genotype and phenotype. The goal of this review is to provide a roadmap to navigate shotgun metagenomic sequencing data and identify new candidate biosynthetic enzymes. We cover both computational and experimental strategies to mine metagenomes and explore protein sequence space with a spotlight on natural product biosynthesis. Specifically, we compare in silico methods for enzyme discovery including phylogenetics, sequence similarity networks, genomic context, 3D structure-based approaches, and machine learning techniques. We also discuss various experimental strategies to test computational predictions including heterologous expression and screening. Finally, we provide an outlook for future directions in the field with an emphasis on meta-omics, single-cell genomics, cell-free expression systems, and sequence-independent methods.
|
|
Scooped by
?
April 22, 2021 12:59 AM
|
Recent progress in synthetic biology allows the construction of dynamic control circuits for metabolic engineering. This technology promises to overcome many challenges encountered in traditional pathway engineering, thanks to their ability to self-regulate gene expression in response to bioreactor perturbations. The central components in these control circuits are metabolite biosensors that read out pathway signals and actuate enzyme expression. However, the construction of metabolite biosensors is laborious and currently a major bottleneck for strain design. Here we present a general method for biosensor design based on multiobjective optimization. Our approach produces libraries of biosensors that optimally trade-off production flux against the genetic burden on the host. We explore properties of control architectures built in the literature, and identify their advantages and caveats in terms of performance and robustness to growth conditions or leaky promoters. We demonstrate the optimality of a control circuit for glucaric acid production in Escherichia coli, which has been shown to increase titer by 2.5-fold as compared to static designs. Our results lay the groundwork for the automated design of control circuits for pathway engineering, with applications in the food, energy and pharmaceutical sectors.
|
|
Scooped by
?
April 21, 2021 2:01 PM
|
The Gram-negative bacterium Vibrio cholerae adapts to changes in the environment by selectively producing the necessary machinery to take up and metabolize available carbohydrates. The import of fructose by the fructose-specific phosphoenolpyruvate (PEP) phosphotransferase system (PTS) is of particular interest because of its putative connection to cholera pathogenesis and persistence. Here, we describe the expression and regulation of fruB, which encodes an EIIA-FPr fusion protein as part of the fructose-specific PTS in V. cholerae. Using a series of transcriptional reporter fusions and additional biochemical and genetic assays, we identified Cra (catabolite repressor/activator) and cAMP receptor protein (CRP) as regulators of fruB expression and determined that this regulation is dependent upon the presence or absence of PTS sugars. Cra (FruR) functions as a repressor, downregulating fruB expression in the absence of fructose when components of PTSFru are not needed. CRP functions as an activator of fruB expression. We also report that Cra and CRP can affect fruB expression independently; however, CRP can modulate cra expression in the presence of fructose and glucose. Evidence from this work provides the foundation for continued investigations into PTSFru and its relationship to the V. cholerae life cycle.
|
|
Scooped by
?
April 21, 2021 1:44 PM
|
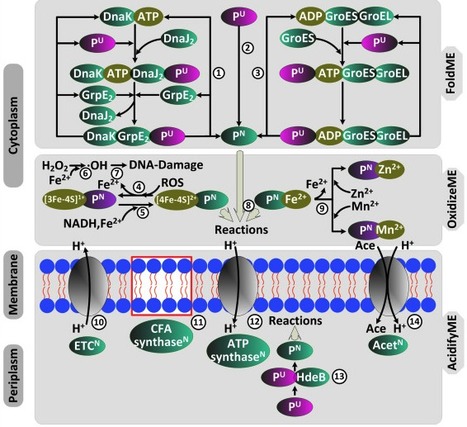
Genome-scale models (GEMs) have been expanded to compute both the metabolic and proteomic states of a cell in different conditions. These multi-scale models called genome-scale models of metabolism and macromolecular expression (ME-models) expand the scope of GEMs and provide greater predictive abilities, and therefore are being increasingly used in systems understanding of microbial biology. In this review, we describe ME-models, their advantages, and computational advances. We then describe extensions of ME-models to model stress response, and a related human red blood cell model that integrate metabolism and macromolecular mechanisms. Finally, we discuss how ME-models were recently used to explain resource allocation tradeoffs for microbial growth in adaptively evolved strains, in synthetic communities, and under stress.
|
|
Scooped by
?
April 21, 2021 11:50 AM
|
Direct determination of RNA structures and interactions in living cells is critical for understanding their functions in normal physiology and disease states. Here, we present PARIS2, a dramatically improved method for RNA duplex determination in vivo with >4000-fold higher efficiency than previous methods. PARIS2 captures ribosome binding sites on mRNAs, reporting translation status on a transcriptome scale. Applying PARIS2 to the U8 snoRNA mutated in the neurological disorder LCC, we discover a network of dynamic RNA structures and interactions which are destabilized by patient mutations. We report the first whole genome structure of enterovirus D68, an RNA virus that causes polio-like symptoms, revealing highly dynamic conformations altered by antiviral drugs and different pathogenic strains. We also discover a replication-associated asymmetry on the (+) and (−) strands of the viral genome. This study establishes a powerful technology for efficient interrogation of the RNA structurome and interactome in human diseases.
|
|
Scooped by
?
April 21, 2021 11:09 AM
|
The opportunistic pathogen Pseudomonas aeruginosa produces an arsenal of virulence factors causing a wide range of diseases in multiple hosts and is difficult to eradicate due to its intrinsic resistance to antibiotics. With the antibacterial pipeline drying up, antivirulence therapy has become an attractive alternative strategy to the traditional use of antibiotics to treat P. aeruginosa infections. To identify P. aeruginosa genes required for virulence in multiple hosts, a random library of Tn5 mutants in strain PAO1-L was previously screened in vitro for those showing pleiotropic effects in the production of virulence phenotypes. Furthermore, the PA4130 isogenic mutant showed substantial attenuation in disease models of Drosophila melanogaster and Caenorhabditis elegans as well as reduced toxicity in human cell lines. Mice infected with this mutant demonstrated an 80% increased survival rate in acute and agar bead lung infection models. PA4130 codes for a protein with homology to nitrite and sulfite reductases. Overexpression of PA4130 in the presence of the siroheme synthase CysG enabled its purification as a soluble protein. Methyl viologen oxidation assays with purified PA4130 showed that this enzyme is a nitrite reductase operating in a ferredoxin-dependent manner. The preference for nitrite and production of ammonium revealed that PA4130 is an ammonia:ferredoxin nitrite reductase and hence was named NirA.
|
|
|
Scooped by
?
April 25, 2021 3:12 PM
|
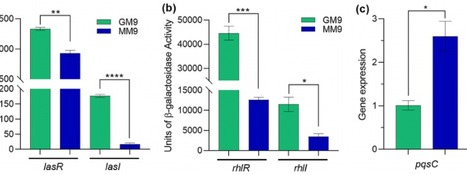
Pseudomonas aeruginosa is an opportunistic pathogen that uses malonate among its many carbon sources. We recently reported that, when grown in blood from trauma patients, P. aeruginosa expression of malonate utilization genes was upregulated. In this study, we explored the role of malonate utilization and its contribution to P. aeruginosa virulence. We grew P. aeruginosa strain PA14 in M9 minimal medium containing malonate (MM9) or glycerol (GM9) as a sole C source and assessed the effect of the growth on quorum sensing, virulence factors, and antibiotic resistance. Growth of PA14 in MM9, compared to GM9, reduced the production of elastases, rhamnolipids, and pyoverdine; enhanced the production of pyocyanin and catalase; and increased its sensitivity to norfloxacin. Growth in MM9 decreased extracellular levels of N‐acylhomoserine lactone autoinducers, an effect likely associated with increased pH of the culture medium; but had little effect on extracellular levels of PQS. At 18 h of growth in MM9, PA14 formed biofilm‐like structures or aggregates that were associated with biomineralization, which was related to increased pH of the culture medium. These results suggest that malonate significantly impacts P. aeruginosa pathogenesis by influencing the quorum sensing systems, the production of virulence factors, biofilm formation, and antibiotic resistance.
|
|
Scooped by
?
April 25, 2021 2:30 PM
|
It has recently been shown that in anaerobic microorganisms the tricarboxylic acid (TCA) cycle, including the seemingly irreversible citrate synthase reaction, can be reversed and used for autotrophic fixation of carbon. This reversed oxidative TCA cycle requires ferredoxin-dependent 2-oxoglutarate synthase instead of the NAD-dependent dehydrogenase as well as extremely high levels of citrate synthase (more than 7% of the proteins in the cell). In this pathway, citrate synthase replaces ATP-citrate lyase of the reductive TCA cycle, which leads to the spending of one ATP-equivalent less per one turn of the cycle. Here we show, using the thermophilic sulfur-reducing deltaproteobacterium Hippea maritima, that this route is driven by high partial pressures of CO2. These high partial pressures are especially important for the removal of the product acetyl-CoA through reductive carboxylation to pyruvate, which is catalysed by pyruvate synthase. The reversed oxidative TCA cycle may have been functioning in autotrophic CO2 fixation in a primordial atmosphere that is assumed to have been rich in CO2.
|
|
Scooped by
?
April 23, 2021 2:37 AM
|
High-throughput directed evolution, implemented in well-controlled in vitro conditions, provides a powerful route for enzyme engineering. Most existing technologies are based on activity screening and require the sequential observation and sorting of each individual variant. By contrast, approaches based on autonomous feedback loops, linking phenotype to genotype replication, enable autonomous selection without screening. However, these approaches are only possible in vivo, or applicable to very specific activities, such as polymerases or ligases. Here, we leverage synthetic molecular networks to create a programmable in vitro feedback loop linking a target enzymatic activity to gene amplification. After encapsulation and lysis of up to 10^7 transformed variants, the genes present in each droplet are amplified according to the activity of the encoded enzyme, resulting in the autonomous enrichment of interesting sequences. Applied to a nicking enzyme with thermal or kinetic selection pressures, this method reveals detailed mutational landscapes and provides improved variants.
|
|
Scooped by
?
April 23, 2021 12:48 AM
|
The Interactive Tree Of Life (https://itol.embl.de) is an online tool for the display, manipulation and annotation of phylogenetic and other trees. It is freely available and open to everyone. iTOL version 5 introduces a completely new tree display engine, together with numerous new features. For example, a new dataset type has been added (MEME motifs), while annotation options have been expanded for several existing ones. Node metadata display options have been extended and now also support non-numerical categorical values, as well as multiple values per node. Direct manual annotation is now available, providing a set of basic drawing and labeling tools, allowing users to draw shapes, labels and other features by hand directly onto the trees. Support for tree and dataset scales has been extended, providing fine control over line and label styles. Unrooted tree displays can now use the equal-daylight algorithm, proving a much greater display clarity. The user account system has been streamlined and expanded with new navigation options and currently handles >1 million trees from >70 000 individual users.
|
|
Scooped by
?
April 22, 2021 11:22 PM
|
Environmental composition is a major, though poorly understood, determinant of microbiome dynamics. Here we ask whether general principles govern how microbial community growth yield and diversity scale with an increasing number of environmental molecules. By assembling hundreds of synthetic consortia in vitro, we find that growth yield can remain constant or increase in a non-additive manner with environmental complexity. Conversely, taxonomic diversity is often much lower than expected. To better understand these deviations, we formulate metrics for epistatic interactions between environments and use them to compare our results to communities simulated with experimentally-parametrized consumer resource models. We find that key metabolic and ecological factors, including species similarity, degree of specialization, and metabolic interactions, modulate the observed non-additivity and govern the response of communities to combinations of resource pools. Our results demonstrate that environmental complexity alone is not sufficient for maintaining community diversity, and provide practical guidance for designing and controlling microbial ecosystems.
|
|
Scooped by
?
April 22, 2021 9:27 PM
|
In this study, some novel plasmids have been constructed for flexible and zero-background molecular cloning, more efficient expression, and purification of proteins with improved strategies. The plasmids pANY4-pL18-ccdB and pANY4-pR18/pL18-ccdB have different promoters in the complementary DNA strands. Therefore, recombinant plasmids for either IPTG-induced or temperature-induced protein expression could be simultaneously constructed in a single molecular cloning process for parallel comparison. Intriguingly, the mutated pL18 and pR18/pL18 promoters performed similar to or even better than the T7 promoter when used for promoting the expression of the GFP or pfLamA enzyme. Moreover, the plasmid pANY8 containing the His-elastin-like polypeptide (ELP)-intein multifunctional tag was constructed, and special purification protocol was designed to obtain purified proteins without the requirement of time-consuming dialysis steps to remove imidazole and high concentration of salt ions. Additionally, the urea-based denaturation and refolding processes can be conveniently integrated into the ELP-mediated precipitation protocol for purification of insoluble inclusion bodies, omitting the time-consuming dialysis steps.
|
|
Scooped by
?
April 22, 2021 2:24 AM
|
We set out to identify phenolic-acid degrading bacteria in forest soils and to evaluate their role in soil priming. Priming occurs when new carbon added to soil stimulates the mineralization of existing carbon stores (Fig. 1). Prior research has shown that phenolics and aromatics could reliably produce soil priming [8, 13], but what caused this effect was unknown. We found that populations of Paraburkholderia were chiefly responsible. We characterized one of the most active species, Paraburkholderia madseniana [14], and named it in honor of our late co-author Dr. Eugene Madsen who initiated our efforts (Fig. 2). P. madseniana is a fast-growing organism that is equipped with diverse and numerous pathways for degrading aromatics. Populations of P. madseniana grew rapidly when either glucose or phenolic acids were added to soil, but priming was specifically induced in the presence of phenolics. Thus, we concluded that Paraburkholderia, and their specialized capacity to degrade phenolics and aromatics, are prime-time players in soil carbon cycling.
|
|
Scooped by
?
April 22, 2021 2:12 AM
|
Genome erosion is a frequently observed result of relaxed selection in insect nutritional symbionts, but it has rarely been studied in defensive mutualisms. Solitary beewolf wasps harbor an actinobacterial symbiont of the genus Streptomyces that provides protection to the developing offspring against pathogenic microorganisms. Here, we characterized the genomic architecture and functional gene content of this culturable symbiont using genomics, transcriptomics, and proteomics in combination with in vitro assays. Despite retaining a large linear chromosome (7.3 Mb), the wasp symbiont accumulated frameshift mutations in more than a third of its protein-coding genes, indicative of incipient genome erosion. Although many of the frameshifted genes were still expressed, the encoded proteins were not detected, indicating post-transcriptional regulation. Most pseudogenization events affected accessory genes, regulators, and transporters, but “Streptomyces philanthi” also experienced mutations in central metabolic pathways, resulting in auxotrophies for biotin, proline, and arginine that were confirmed experimentally in axenic culture. In contrast to the strong A+T bias in the genomes of most obligate symbionts, we observed a significant G+C enrichment in regions likely experiencing reduced selection. Differential expression analyses revealed that—compared to in vitro symbiont cultures—“S. philanthi” in beewolf antennae showed overexpression of genes for antibiotic biosynthesis, the uptake of host-provided nutrients and the metabolism of building blocks required for antibiotic production. Our results show unusual traits in the early stage of genome erosion in a defensive symbiont and suggest tight integration of host–symbiont metabolic pathways that effectively grants the host control over the antimicrobial activity of its bacterial partner.
|
|
Scooped by
?
April 21, 2021 3:54 PM
|
The intimate interactions of indigenous crops with their associated microbiomes during long-term co-evolution strengthen the capacity and flexibility of crops to cope with biotic and abiotic stresses. This represents a promising untapped field for searching novel tools to sustainably increase crop productivity. However, the current capability of harnessing the power of indigenous crop microbiomes for sustainable crop production is limited due to low efficiency of separating the targeted functional microbes. Here, we highlight the potential benefits and existing challenges of utilizing indigenous crop microbiomes to reduce agrochemical inputs and increase crop resistance to biotic and abiotic stresses. We propose a framework using Raman-spectroscopy-based single-cell-sorting technology combined with a synthetic community approach to design and optimize a functionally reliable ‘beneficial biome’ under controlled conditions. This framework will offer opportunities for sustainable agriculture and provide a new direction for future studies. Crop microbiomes provide plants with beneficial functions including increased nutrient acquisition and stress tolerance, but the current capability of utilizing indigenous crop microbiomes is limited due to low efficiency of separating the targeted functional microbes. A newly proposed framework using single-cell-sorting Raman spectroscopy combined with a synthetic community approach has the potential to design and optimize a ‘beneficial biome’.
|
|
Scooped by
?
April 21, 2021 1:54 PM
|
Computational modeling of microbial communities using GEnome-scale Models (GEMs) of metabolism is a new frontier in systems biology. Here, we discuss recent developments in this area ranging from high-throughput GEMs reconstruction pipelines to approaches for modeling under steady-state and for simulating temporal, evolutionary and spatiotemporal dynamics of microbial communities. We categorize these approaches based on Flux Balance Analysis or Elementary Mode Analysis of mixed-bag and compartmentalized GEMs and discuss their scope of applications and scalability for large-scale simulations. Additionally, we review computational tools using GEMs for the design of microbial communities and recent efforts to integrate GEMs and machine learning for predicting inter-species interactions. We conclude with discussing best practices for using these tools and potential avenues for future developments.
|
|
Scooped by
?
April 21, 2021 12:10 PM
|
Antibiotic resistance (AR) is a threat to modern medicine, and plasmids are driving the global spread of AR by horizontal gene transfer across microbiomes and environments. Determining the mobile resistome responsible for this spread of AR among environments is essential in our efforts to attenuate the current crisis. Biosolids are a wastewater treatment plant (WWTP) byproduct used globally as fertilizer in agriculture. Here, we investigated the mobile resistome of biosolids that are used as fertilizer. This was done by capturing resistance plasmids that can transfer to human pathogens and commensal bacteria. We used a higher-throughput version of the exogenous plasmid isolation approach by mixing several ESKAPE pathogens and a commensal Escherichia coli with biosolids and screening for newly acquired resistance to about 10 antibiotics in these strains. Six unique resistance plasmids transferred to Salmonella typhimurium, Klebsiella aerogenes, and E. coli. All the plasmids were self-transferable and carried 3–6 antibiotic resistance genes (ARG) conferring resistance to 2–4 antibiotic classes. These plasmids-borne resistance genes were further embedded in genetic elements promoting intracellular recombination (i.e., transposons or class 1 integrons). The plasmids belonged to the broad-host-range plasmid (BHR) groups IncP-1 or PromA. Several of them were persistent in their new hosts when grown in the absence of antibiotics, suggesting that the newly acquired drug resistance traits would be sustained over time. This study highlights the role of BHRs in the spread of ARG between environmental bacteria and human pathogens and commensals, where they may persist. The work further emphasizes biosolids as potential vehicles of highly mobile plasmid-borne antibiotic resistance genes.
|
|
Scooped by
?
April 21, 2021 11:32 AM
|
Base editing is a powerful genome editing approach that enables single-nucleotide changes without double-stranded DNA breaks (DSBs). However, off-target effects as well as other undesired editings at on-target sites remain obstacles for its application. Here, we report that bubble hairpin single guide RNAs (BH-sgRNAs), which contain a hairpin structure with a bubble region on the 5′ end of the guide sequence, can be efficiently applied to both cytosine base editor (CBE) and adenine base editor (ABE) and significantly decrease off-target editing without sacrificing on-target editing efficiency. Meanwhile, such a design also improves the purity of C-to-T conversions induced by base editor 3 (BE3) at on-target sites. Our results present a distinctive and effective strategy to improve the specificity of base editing.
|