Your new post is loading...

|
Scooped by
?
Today, 1:44 AM
|
Conjugative plasmids drive bacterial evolution and niche adaptation. Their carriage often corresponds with changes to the host transcriptome, but it is unknown during which stage of the conjugation process these transcriptional changes are initiated, and how gene transfer is modulated as they occur. This is because they are normally studied after their full acquisition by conjugation has already occurred, not during the process. Here, active RP4 conjugation revealed that the transcriptional response to plasmid acquisition is almost immediate and is host and surface factor dependent. Responses included activation of non-SOS stress pathways, motility, exopolysaccharide production, anaerobic respiration, and metabolic adaptation. These responses were not a barrier to conjugation, indicating that their significance relates to host homeostasis rather than gene transfer. The recipient could prevent these responses by blocking conjugation through capsule expression. RP4 expression was also inhibited by recipient capsule, since capsule physically blocked donor-recipient contact. Unexpectedly, within a population, RP4 was found to exploit single cell variation in recipient capsule thickness and to successfully conjugate with recipient bacteria with reduced capsule. These results show a novel interplay between plasmid and surface architecture across diverse species that controls the conjugation transcriptional landscape, with important ramifications for plasmid dissemination and evolution.
|
|
Scooped by
?
Today, 1:09 AM
|
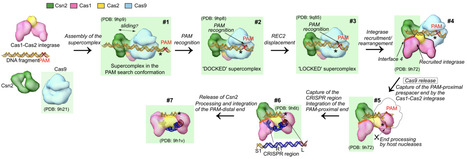
During CRISPR-Cas adaptation, prokaryotic cells become immunized by the insertion of foreign DNA fragments, termed spacers, into the host genome to serve as templates for RNA-guided immunity. Spacer acquisition relies on the Cas1-Cas2 integrase and accessory proteins like Cas4, which select DNA sequences flanked by the protospacer adjacent motif (PAM) and insert them into the CRISPR array. It has been shown that in type II-A systems selection of PAM-proximal prespacers is mediated by the effector nuclease Cas9, which forms a 'supercomplex' with the Cas1-Cas2 integrase and the Csn2 protein. However, the supercomplex structure and the role of the ring-like Csn2 protein remain unknown. Here, we present cryo-electron microscopy structures of the type II-A prespacer selection supercomplex in the DNA-scanning and two different PAM-bound configurations. Our study uncovers the mechanism of Cas9-mediated prespacer selection in type II-A CRISPR-Cas systems, and reveals the role of the accessory protein Csn2, which serves as a platform for the assembly of Cas9 and Cas1-Cas2 integrase on prespacer DNA, reminiscent of the sliding clamp in DNA replication. Repurposing of Cas9 by the CRISPR adaptation machinery for prespacer selection characterized here demonstrates Cas9 plasticity and expands our knowledge of the Cas9 biology.
|
|
Scooped by
?
Today, 12:54 AM
|
Regulatory elements are essential components of plant genomes that have shaped the domestication and improvement of modern crops. However, their identity, function and diversity remain poorly characterized, limiting our ability to harness their full power for agricultural advances using induced or natural variation. Here we mapped transcription factor (TF) binding for 200 TFs from 30 families in two distinct maize inbred lines historically used in maize breeding. TF binding comparison revealed widespread differences between inbreds, driven largely by structural variation, that correlated with gene expression changes and explained complex quantitative trait loci such as Vgt1, an important determinant of flowering time, and DICE, an herbivore resistance enhancer. CRISPR–Cas9 editing of TF binding regions validated the function and structure of regulatory regions at various loci controlling plant architecture and biotic resistance. Our maize TF binding catalogue identifies functional regulatory regions and enables collective and comparative analysis, highlighting its value for agricultural improvement. Genome-wide comparative analysis of DNA binding for 200 transcription factors in two maize inbred lines reveals prevalent genotype-specific variation that is associated with gene expression differences and agronomic traits.
|
|
Scooped by
?
June 12, 11:56 PM
|
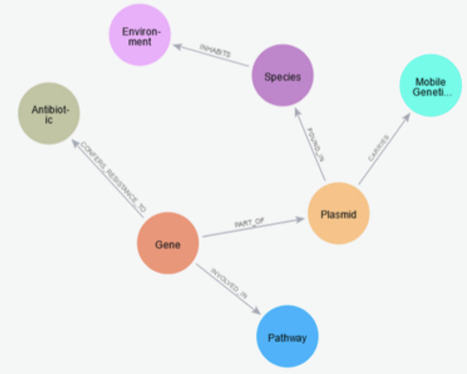
Horizontal gene transfer (HGT) is a key mechanism driving the rapid dissemination of antimicrobial resistance (AMR) among bacterial populations, posing a severe threat to global public health. Traditional detection methods for HGT, such as phylogenetic incongruence and compositional analysis, are often limited in their scope and accuracy. To address these challenges, we propose a comprehensive knowledge graph (KG)-based framework that integrates genomic, environmental, biochemical, and ontological datasets to detect and analyze HGT events contributing to AMR. Our KG captures relationships among genes, plasmids, species, mobile genetic elements, antibiotics, and ecological factors using curated sources like NCBI, CARD, ICEberg, MGnify, and KEGG. We enrich nodes and edges with biological (e.g., GC content, codon bias) and chemical (e.g., binding affinities, environmental exposure) signatures and apply ontology-based semantic enrichment using ARO and GO. A Graph Neural Network (GNN) trained on the KG achieves high accuracy (92%) and F1-score (0.90) in predicting HGT events, outperforming conventional methods. Community detection identifies AMR dissemination hubs, while environmental analyses reveal strong correlations between antibiotic concentrations and resistance gene prevalence. Our framework demonstrates strong biological and chemical validation and provides a scalable, interpretable, and data-rich tool for AMR surveillance, with implications for clinical, agricultural, and environmental microbiology.
|
|
Scooped by
?
June 12, 11:21 PM
|
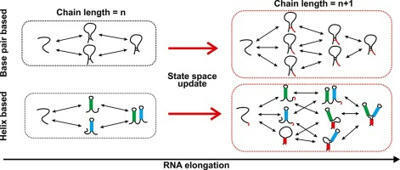
An RNA folds as it is transcribed. RNA folding during transcription differs fundamentally from thermodynamic folding. While thermodynamic folding reaches an equilibrium ensemble of structures, cotranscriptional folding is a kinetic process where the RNA structure evolves as the chain elongates during transcription. This dynamic folding pathway causes cotranscriptional structures often to deviate from thermodynamic predictions, as the system rarely reaches equilibrium. Since these cotranscriptional effects can persist in the mature RNA's structure, understanding this kinetic process is crucial. While experimental studies of cotranscriptional folding have been successful, they remain resource-intensive. Computational modeling has emerged as an increasingly practical and powerful approach for investigating these dynamics. This short review examines current computational methods and tools for simulating cotranscriptional folding, with the goal of advancing our understanding of RNA folding mechanisms.
|
|
Scooped by
?
June 12, 11:00 PM
|
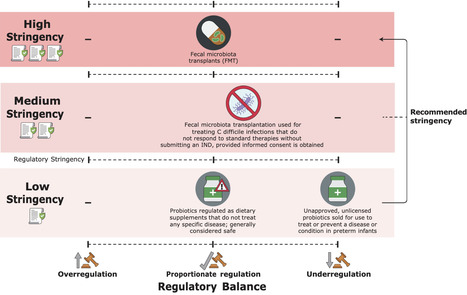
The three main types of live bacterial therapies – probiotics, fecal/microbiome transplants, and engineered bacterial therapies – hold immense potential to revolutionize medicine. While offering targeted and personalized treatments for various diseases, these therapies also carry risks such as adverse immune reactions, antibiotic resistance, and the potential for unintended consequences. Therefore, developing and deploying these therapies necessitates a robust regulatory framework to protect public health while fostering innovation. In this paper, we propose a novel conceptual tool – the Ladder of Regulatory Stringency and Balance—which can assist in the design of robust regulatory regimes which encompass medicine practices based not only on definitive Randomized Controlled Trials (RCTs), but also on meta-analyses, observational studies, and clinicians experience. Regulatory stringency refers to the strictness of regulations, while regulatory balance concerns the degree of alignment between the regulatory framework governing a technology and the actual risks posed by specific products within that technology. Focusing on the US regulatory environment, we subsequently position the three types of live bacterial therapies on the Ladder. The insight gained from this exercise demonstrates that probiotics are generally positioned at the bottom of the Ladder, corresponding to low-stringency regulation, with a proportionate regulatory balance. However, probiotics intended for high-risk populations are currently subject to low-stringency regulations, resulting in under-regulation. Our analysis also supports the conclusion that fecal microbiota transplants (FMT) for recurrent Clostridium difficile infection should be positioned close to but below the threshold for under regulation by the U.S. Food and Drug Administration (FDA), and we recommend improved donor screening procedures, preservation and processing, storage, and distribution. Our framework can serve as a scale to assess regulatory gaps for live bacterial therapies and to identify potential solutions where such gaps exist.
|
|
Scooped by
?
June 12, 10:43 PM
|
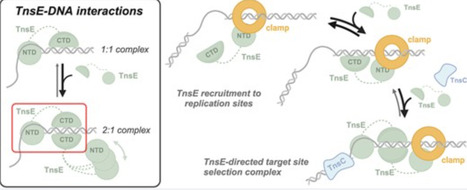
Tn7 transposable elements are known for their sophisticated target-site selection mechanisms. For the prototypical Tn7 element, dedicated transposon-encoded proteins direct insertions to either a conserved site in the chromosome or replicating DNA structures in conjugal plasmids, ensuring the vertical and horizontal spread of the element. While the pathway targeting the attTn7 site in the bacterial chromosome has been extensively studied, the pathway targeting DNA replication structures remains poorly understood. We have used an integrative structural biology approach to elucidate how the Tn7-encoded protein TnsE recognizes replication sites. Using native mass spectrometry, we found that TnsE forms 1:1 and 2:1 (TnsE:DNA) complexes on 3′-recessed DNA, with gain-of-function TnsE variants favoring the formation of 2:1 complexes. Structural characterization confirms that two TnsE molecules bind to DNA with the C-terminal domain of the protein recognizing duplex DNA, leaving the N-terminal domain to impose DNA substrate specificity and recruit the core transposition machinery. Collectively, our work is consistent with a model where TnsE-mediated target-site selection relies on the formation of an asymmetric TnsE:DNA complex to recruit the Tn7 transposase to DNA replication structures.
|
|
Scooped by
?
June 12, 4:33 PM
|
Functional analysis of bacterial genes or genomic fragments in vivo primarily relies on the analysis of knockout strains. Although various methods have successfully generated bacterial knockout mutants, the parallel operation of multiple sites, especially in biofoundries, remains challenging. New technological refinements of existing methods are necessary for high-throughput genomic editing in bacteria. In this study, to modify numerous sites in parallel, we optimized the linear donor DNA by adding modification at the different positions and achieved high-efficiency recombination with chemical transformation. Then, by combining with the CRISPR system, we established a guide sequence-independent and donor DNA-mediated genomic editing (GIDGE) method, enabling efficient and scarless engineering of common E. coli strains as well as wild-type strains such as E. coli MG1655, with particularly marked advantages demonstrated in E. coli Nissle 1917. This method allows for high-throughput genomic engineering in a 96-well format and is useful for sequence deletion with a wide range of lengths, sequence insertion, sequence replacement, and point mutation. As a proof-of-concept study, we constructed 96 single-gene knockout mutants and a genomic large-fragment deletion library in E. coli K-12 MG1655 using the GIDGE method. This high-throughput and easy-to-use method has great potential for automation and can be adapted for use in biofoundries.
|
|
Scooped by
?
June 12, 4:17 PM
|
Using modern genomic tools, Feng et al. revisited Mendel’s seven pea traits in a recent Nature study, uncovering the molecular genetic basis of all of them, including the three unresolved ones: pod color, pod shape, and flower position. Their work highlights the level of complexity provided by structural variation that could impact genes and their regulatory regions and thus influence the expression of plant traits. The authors demonstrate how revisiting foundational experiments with contemporary tools can manifest novel biological insights and also guide future crop improvement.
|
|
Scooped by
?
June 12, 4:02 PM
|
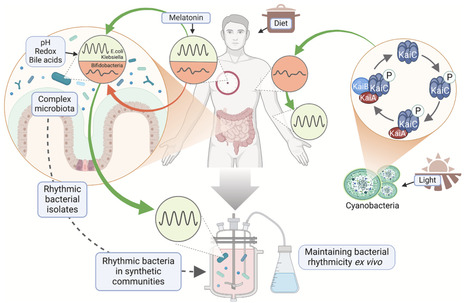
Daily oscillations in microbiota composition and function are emerging as an important element in host-microbiota interactions. Here, we summarize features of the microbiota that undergo diurnal rhythms, their development, their impact on the biology of the host, and their relevance to human health and disease. In particular, we focus on the intrinsic and extrinsic factors that regulate microbiota oscillations and the multifaceted roles that microbiota rhythmicity plays in host physiology, immunity, and metabolism. Given the pervasive impact of intestinal microorganisms on host health, understanding the origins and functions of microbiota rhythms is a critical aspect of the circadian biology of the meta-organism.
|
|
Scooped by
?
June 12, 3:50 PM
|
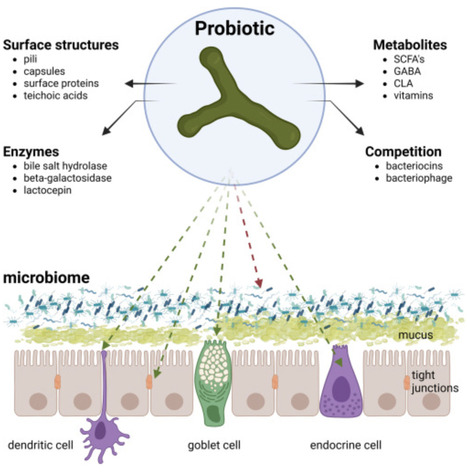
Probiotics, prebiotics, synbiotics, and postbiotics are required, by definition, to confer a health benefit on the host. It is often presumed the host microbiome plays a central role in the mechanism of action of these substances, collectively referred to here as “biotics.” However, the definitions of both probiotics and postbiotics do not include an associated mechanism nor the involvement of the microbiome. The definitions of prebiotics and synbiotics require evidence of selective utilization by the host microbiome, but do not state that confirmatory evidence must be provided that this utilization causes the associated health benefit. In this perspective, we discuss evidence supporting a role for the microbiome in delivering these health benefits and whether or not measuring microbiome alterations can serve as important readouts of efficacy. We also discuss the possibility of expanding the biotics family with substances such as bacteriophage, fermented foods, and live dietary microbes.
|
|
Scooped by
?
June 12, 1:17 AM
|
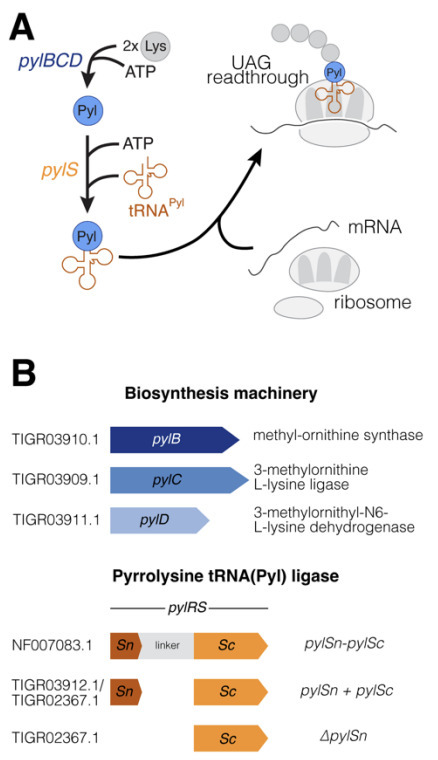
Natural genetic code expansion is a phenomenon wherein an additional amino acid is encoded by a stop codon. These non-standard amino acids are beneficial as they facilitate novel biochemical reactions. However, code expansion leads to ambiguity at the recoded stop codon, which can either be read through or terminated. Pyrrolysine (Pyl) is encoded by the amber codon (TAG/UAG) and is widespread in archaea, where it is required for methylamine-mediated methanogenesis, an environmentally important metabolism. Mechanisms to conditionally suppress the amber stop codon for Pyl installation during protein synthesis have not been identified. Using the model methanogen, Methanosarcina acetivorans, we demonstrate that Pyl-encoding archaea maintain an ambiguous genetic code wherein UAG encodes dual meaning as stop and Pyl. Our data suggest that expression of Pyl biosynthesis and incorporation genes is tuned to the cellular demand for Pyl, which allows these archaea to navigate ambiguous stop decoding in response to environmental cues.
|
|
Scooped by
?
June 12, 12:29 AM
|
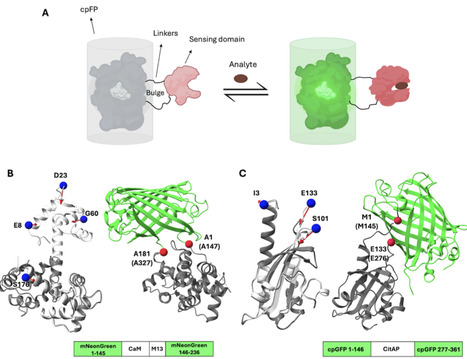
Proteins exhibit remarkable conformational flexibility, enabling precise functional regulation through allostery. A key application of allostery is in the design of protein-based sensors, which detect environmental changes—such as ligand binding or post-translational modifications—and convert these cues into measurable signals (e.g., fluorescence). Here, we investigate a series of ligand-binding proteins that serve as sensing domains in direct-response fluorescent biosensors, wherein ligand binding enhances fluorescence output. We employ a multiple force application approach which we term Multiply Perturbed Response (MPR) to identify “hot spot” residues that drive the conformational transition from an apo (inactive/OFF) to a holo (active/ON) state. We first present two efficient computational approaches to determine residues and forces that maximize the overlap of the observed conformational change. We then determine the overlap maximizer residues for up to five force insertion locations, and we compare them with actual insertion sites used in existing biosensors. Our analysis shows that the allosteric residues identified by MPR coincide with the fluorescent-protein insertion sites that were mapped experimentally through extensive trial-and-error. This work enhances the utility of linear response theory-based methods in uncovering multiple functionally significant regions that trigger a known conformational change. While the validity of the harmonic approximation in anharmonic conformational transitions needs additional validation, MPR gives a good starting point to explore allosteric sites. The approach might prove useful not only in the design of biosensors, but may also find applications in offering physics-based collective variables in mapping conformational transition pathways of proteins.
|
|
|
Scooped by
?
Today, 1:42 AM
|
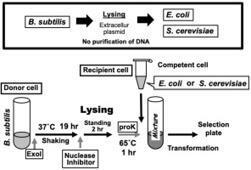
Purified DNA plasmids traditionally used for microbial transformation have been supplanted by extracellular plasmids released via host bacterial lysis, offering an alternative approach for DNA-plasmid delivery. Specifically, shuttle vector plasmids liberated from host Bacillus subtilis were directly employed for the transformation of chemically competent cells Escherichia coli, eliminating the need for biochemical purification. This unconventional DNA delivery technique, referred to as ’Cell Lysis Technology to provide Transformable Extra-cellular DNA; CELyTED’, has been successfully adapted for the transformation of microorganism Saccharomyces cerevisiae as well. The protocol includes optimized conditions for efficient cell lysis of the donor host cells. Notably, ’ CELyTED ’ enables the introduction of large-sized DNA plasmids exceeding 50 kb into target microorganisms mitigating the potential adverse effects of physical shearing during the purification process. This simplicity in the delivery protocol makes it versatile for both prokaryotic and eukaryotic microorganisms, establishing a fundamental platform in the synthetic genome field. Our study demonstrates the feasibility of introducing large DNA plasmids into cells E. coli and S. cerevisiae using the lysate of donor host cells, showcasing the potential of ‘CELyTED ’ as a streamlined approach in genetic transformation methodologies.
|
|
Scooped by
?
Today, 1:01 AM
|
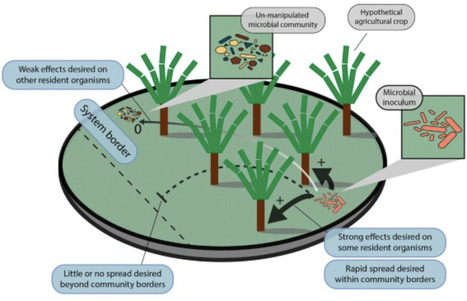
Microbial inoculants are increasingly used for beneficial purposes in agriculture, bioremediation, and medicine, but they can carry risks of generating invasive microbes. Here, we present a roadmap for guarding against these invasions, proposing developing (i) coherent mechanistic understandings of how microbial inoculants can effect invasions, (ii) predictive models forecasting microbial invasion risks, and (iii) effective management strategies. To guide mechanistic understandings, we distill 17 guiding hypotheses. For predictive modeling, we highlight data collection needs and qualitative approaches. For management strategies, we stress the importance of accurately weighing the risks against benefits. The unified approach presented here provides a route toward an effective research and management infrastructure for microbial inoculants in order to avoid potentially catastrophic microbial invasions.
|
|
Scooped by
?
Today, 12:09 AM
|
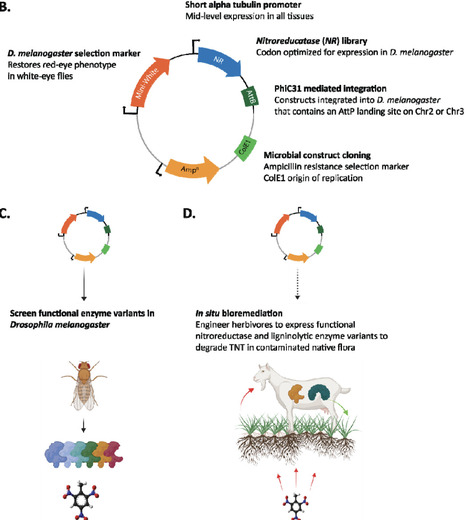
TNT (2,4,6-trinitrotoluene) from unexploded ordinances is a common environmental pollutant near munitions factories, military training sites, and areas of armed conflict. As physical and chemical approaches for TNT remediation are costly and difficult to scale, in situ bioremediation is an attractive alternative. TNT is a phytoaccumulative pollutant. Herbivores engineered to detoxify TNT are a potentially cost-effective platform to bioremediate large areas of contaminated sites while grazing or browsing. As a first step towards engineering herbivores with metabolic capabilities for TNT degradation, we engineered the animal genetic model, Drosophila melanogaster, to screen a library of microbial nitroreductases that can catalyse the initial degradation steps required for TNT degradation. We find strong, cofactor-dependent activity in fly lysates engineered to express NsfA from Escherichia coli, and NsfI from Enterobacter cloacae. bioremediation
|
|
Scooped by
?
June 12, 11:41 PM
|
Cas1 and Cas2 are the hallmark proteins of prokaryotic adaptive immunity. However, these two proteins are often fused to other proteins and the functional association of these fusions often remain poorly understood. Here we purify Cas1 and the Cas2/3 fusion protein from Pseudomonas aeruginosa. We determine multiple structures of the Cas1-2/3 complex at distinct stages of CRISPR adaptation. Collectively, these structures reveal a prominent, positively charged channel on one face of the integration complex that captures short fragments of foreign DNA. Foreign DNA binding triggers conformational changes in Cas2/3 that expose new DNA binding surfaces necessary for homing the DNA-bound integrase to specific CRISPR loci. The length of the foreign DNA substrate determines if Cas1-2/3 docks completely onto the CRISPR repeat to successfully catalyze two sequential transesterification reactions required for integration. Taken together, these structures clarify how the Cas1-2/3 proteins orchestrate foreign DNA capture, site-specific delivery, and integration of new DNA into the bacterial genome.
|
|
Scooped by
?
June 12, 11:03 PM
|
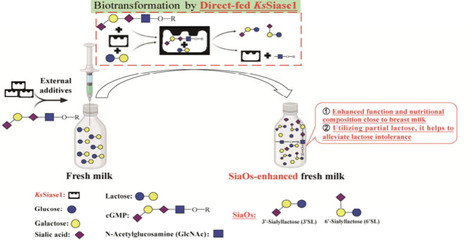
Sialyloligosaccharides, critical components of human milk oligosaccharides, are pivotal for infant immunity and development. Recent advances have identified sialidases with trans-sialylation activity as promising biocatalysts for cost-effective sialyloligosaccharide synthesis using economical substrates. Herein, we report a novel sialidase (KsSiase1) from Kribbella sp. ALI-6-A with enhanced catalytic properties and broad substrate specificity, showing preferential hydrolysis: α2,3 > α2,6 > α2,8. The enzyme demonstrated robust trans-sialidase activity, achieving 3.40 g/L sialyloligosaccharide production using 0.5 U/mL KsSiase1, 6% lactose, and 8% cGMP. Implementing a direct-fed enzymatic strategy in raw milk with 1 U/mL KsSiase1 and 6% cGMP, we achieved in situ sialyloligosaccharide enrichment (2.49 g/L) while concomitantly reducing lactose. This biocatalytic system establishes a sustainable, scalable platform for functional food production, merging lactose valorization with sialyloligosaccharides biosynthesis through enzyme–substrate adaptability, addressing both industrial demands and nutritional needs in infant formula development.
|
|
Scooped by
?
June 12, 10:53 PM
|
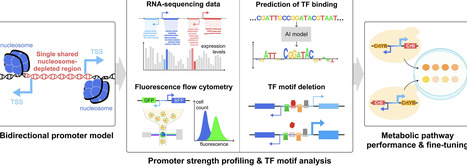
Bidirectional promoters (BDPs) hold great promise for applications in synthetic biology by enabling co-expression of multiple genes with minimized promoter size. However, the lack of well-characterized BDPs along with an incomplete understanding of their regulatory mechanisms limits broader applications. Here, we conducted genome-wide screening and characterization of 749 BDP candidates containing a single shared nucleosome-depleted region in yeast Saccharomyces cerevisiae. A pronounced asymmetry in BDP strength was observed using both transcriptomic and fluorescence reporter analyses. We demonstrated that these unbalanced BDP strengths could be utilized for fine-tuning metabolic flux in yeast, achieving yields comparable to or exceeding those of commonly used constitutive or inducible promoters for terpenoid production under the examined conditions. Using in silico mutagenesis guided by the DREAM-CNN yeast cis-regulatory AI prediction model, we identified conserved activator-binding hotspots within the central region of 63.8% of identified BDP candidates. Disruption of these hotspots in six selected BDPs significantly reduced promoter strength in both orientations, suggesting that these AI-predicted motifs are indeed critical for the functionality of BDPs. Overall, this study provides a comprehensive framework for BDP identification and engineering, leveraging AI-guided models to advance rational synthetic promoter design, thus paving the way for precise genetic control in synthetic biology.
|
|
Scooped by
?
June 12, 4:37 PM
|
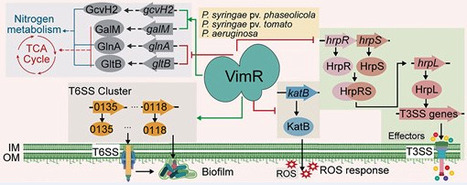
Pseudomonas syringae, a highly destructive plant bacterial pathogen causing severe disease and significant yield losses in agriculture globally, has complex regulatory systems involving many transcriptional factors (TFs). Although the LysR-type transcriptional regulator (LTTR) protein family is a well-known group of TFs involved in diverse physiological functions, the roles of LTTRs in P. syringae remain largely unknown. In this study, we characterized a LysR-type TF, PSPPH4638, and designated it as the virulence and metabolism regulator VimR. Genome-wide identification of VimR using chromatin immunoprecipitation sequencing revealed 1032 binding sites in the genome, of which 85% were in intergenic regions. Transcriptomic analysis showed altered expression of 454 and 82 genes in response to ΔvimR in King’s B medium (KB) and minimal medium (MM), respectively. Conjoint analysis showed that 99 genes were directly affected by VimR in KB. VimR was identified as a repressor of the type III secretion system, oxidative stress response, and key metabolic pathways such as the tricarboxylic acid cycle. In addition, we found that VimR was positively involved in the type VI secretion system and alanine, aspartate, and glutamate metabolism. Further verification showed that VimR was widely present in Pseudomonas, displaying similar binding capacity in different strains of P. syringae, and similar regulatory functions in Pseudomonas aeruginosa. Taken together, our findings identified a conserved master TF that regulates type III secretion system, type VI secretion system, and multiple metabolic pathways in Pseudomonas.
|
|
Scooped by
?
June 12, 4:20 PM
|
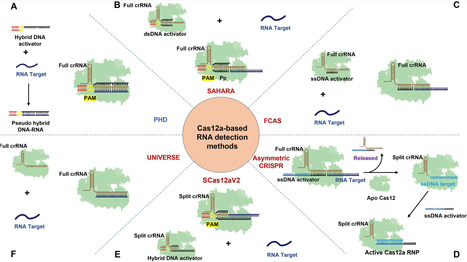
The CRISPR/Cas12a system is known for its intrinsic RNA-guided trans-cleavage activity; however, its RNA detection sensitivity is limited, with conventional methods typically achieving detection limits in the nanomolar range. Here, we report the development of a “pseudo hybrid DNA–RNA” (PHD) assay that significantly enhances the RNA detection capability of Cas12a. The PHD assay achieves a striking detection limit of 7.7 pM using single CRISPR RNA (crRNA) and 33.8 fM using pooled crRNAs. Importantly, this assay exhibits ultra-high specificity, capable of distinguishing mutated RNA target sequences at the protospacer adjacent motif (PAM)-distal region. It can also detect ultrashort RNA sequences as short as 6–8 nt and long RNAs with complex secondary structures. Additionally, the PHD assay enables PAM-free attomolar-level DNA detection. We further demonstrate the practical utility of the PHD assay by successfully detecting miR-155 biomarkers and human pappilloma virus 16 DNA in clinical samples. We anticipate that the design principles established in this study can be extended to other CRISPR/Cas enzymes, thereby accelerating the development of powerful nucleic acid testing tools for various applications.
|
|
Scooped by
?
June 12, 4:16 PM
|
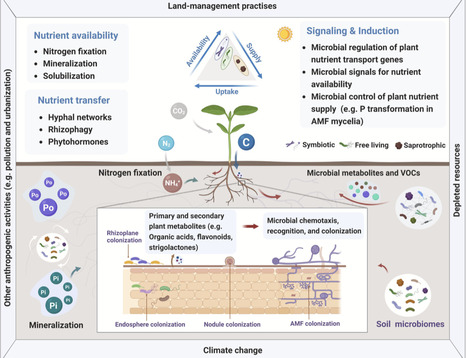
Plants and microbiomes have co-evolved for millennia. Through this co-evolution, microbiomes have become essential for plant nutrient acquisition, which involves plant signaling, microbial sensing, and acquiring and sharing nutrients. In this review, we synthesize recent advancements in the complex associations of molecular, physiological, and eco-evolutionary mechanisms that underpin microbe-facilitated plant nutrient uptake. Focusing on emerging insights in plant-microbial communication and metabolic pathways, we evaluate potential opportunities to harness plant microbiomes to sustainably supply nutrients in agricultural and natural ecosystems. However, further progress is constrained by key knowledge gaps. We propose an amended conceptual framework for advancement that includes a holistic understanding of eco-evolutionary relationships with explicit consideration of signaling and sensing mechanisms. Finally, we argue that advancing fundamental science by utilizing emerging analytical approaches in an integrated way is critical to develop effective microbiome-informed tools that can enhance plant nutrient acquisition and promote long-term food security and environmental sustainability.
|
|
Scooped by
?
June 12, 3:57 PM
|
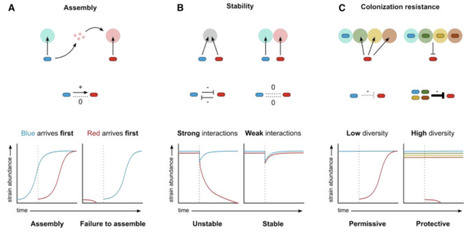
Many plants and animals, including humans, host diverse communities of microbes that provide many benefits. A key challenge in understanding microbiomes is that the species composition often differs among individuals, which can thwart generalization. Here, we argue that the key to identifying general principles for microbiome science lies in microbial metabolism. In the human microbiome and in other systems, every microbial species must find ways to harvest nutrients to thrive. The available nutrients in a microbiome interact with microbial metabolism to define which species have the potential to persist in a host. The resulting nutrient competition shapes other mechanisms, including bacterial warfare and cross-feeding, to define microbiome composition and properties. We discuss impacts on ecological stability, colonization resistance, nutrient provision for the host, and evolution. A focus on the metabolic ecology of microbiomes offers a powerful way to understand and engineer microbiomes in health, agriculture, and the environment.
|
|
Scooped by
?
June 12, 3:36 PM
|
Agricultural long-term field trials provide fundamental data on crop performance and soil characteristics under diverse management practices. This information represents essential knowledge for upcoming challenges in food and nutrition security. Data provided here have been compiled since 2004 from a nitrogen(N)-fertilization intensity, tillage, and crop rotation field trial in Central Germany including standardized metrics regarding soil management, physical soil properties, crop management, crop characteristics, yield, and harvest quality parameters. In 2015, the field trial became a member of the German Agricultural Soil Research Program BonaRes. Numerous measurement results were added including plant physiology and soil and rhizosphere microbiology. DNA of bacterial/archaeal and fungal microbiomes was sequenced in the rhizosphere and root-associated soil following a meta-barcoding approach. Taxonomic and relative abundance data were included in the dataset. The dataset is the first to include information on root characteristics, soil and rhizosphere microbiomes, and crop gene expression. We encourage reuse of these biological field trial data in terms of meta-analysis, modeling and AI approaches.
|
|
Scooped by
?
June 12, 12:38 AM
|
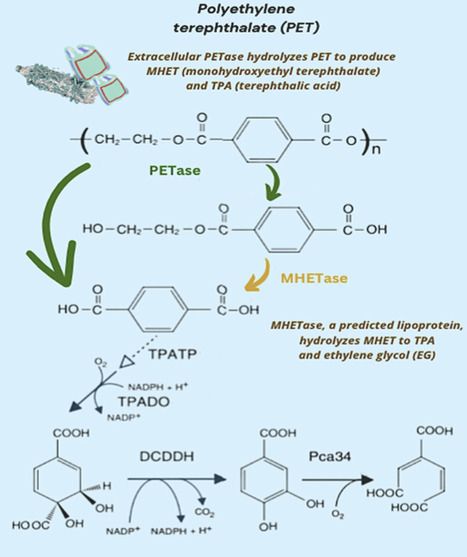
Additives such as stabilizers, plasticizers, and fillers are commonly used in relatively small amounts to enhance the structure of plastics. Notably, some of these additives, including moieties of compounds containing nitrogen, phosphorus, and sulfur, are essential for microbial proliferation. Most studies on plastic degradation have primarily focused on the potential of microorganisms to assimilate carbon from plastics to support their growth, a strategy that has yet to yield significant success. However, studies investigating the removal of non‑carbon moieties of additives from plastics, which could weaken their structure and thereby enhance fragmentation, remain largely unexplored. This review highlights the potential of harnessing microbial processes that target the non‑carbon moieties of additives to weaken the structural integrity of plastics. The weakened plastic may then become more accessible to heterotrophic microbes, thereby accelerating its degradation.
|