 Your new post is loading...

|
Scooped by
mhryu@live.com
Today, 2:18 AM
|
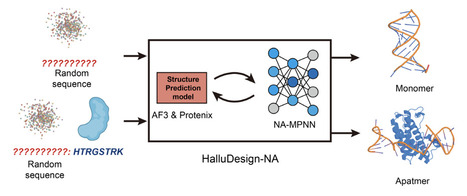
AlphaFold3 has revolutionized the prediction of biomolecular structures and interactions, including atomic-level modeling of nucleic acids. However, the de novo design of structured and functional nucleic acids remains a significant challenge. Here, we extend our HalluDesign framework to nucleic acid design by integrating NA-MPNN for nucleic acid sequence optimization and design. This new framework, HalluDesign-NA, enables iterative sequence-structure co-optimization, facilitating the de novo design of nucleic acids. Computational benchmarking across ssDNA, ssRNA, and aptamer design tasks demonstrates consistent improvements in confidence scores (pLDDT, ipTM), supporting the feasibility of de novo nucleic acid design under various constraints, such as sequence length, symmetry, and protein structure context. We anticipate that HalluDesign-NA will accelerate the de novo design of functional nucleic acids for applications in biotechnology and medicine. The source code for HalluDesign-NA is available at https://github.com/MinchaoFang/HalluDesign_NA.
|
|
Scooped by
mhryu@live.com
Today, 2:08 AM
|
Polyhydroxyalkanoates (PHA), a class of biodegradable polyesters synthesized by microorganisms, have emerged as a key platform for advanced antimicrobial biomedicine. Distinguished by their exceptional biocompatibility, tunable degradation kinetics and versatile material properties, PHA offer a sustainable, functional alternative to conventional polymers. Furthermore, specific PHA materials exhibit inherent antimicrobial properties, opening new avenues for the developing eco-efficient medical materials. This review systematically outlines the multifaceted antibacterial mechanisms of PHA, including their intrinsic structure-activity relationships, the bioactive roles of their degradation products, and their effectiveness as carriers for engineered antimicrobial agents. Recent advances in PHA-based applications are also critically summarized, with a focus on wound dressings, surgical sutures, orthopaedical implants, and targeted drug delivery systems. The therapeutic performance of these applications in infection control and tissue repair is also highlighted. Finally, this paper discusses the current challenges to translation and propose strategic research directions, aiming to chart a developmental roadmap for the next generation of intelligent, PHA-based, antimicrobial solutions.
|
|
Scooped by
mhryu@live.com
Today, 1:36 AM
|
DNA sliding clamps are central coordinators of genome replication and maintenance, yet the full binding network (“interactome”) of the bacterial β-clamp remains incompletely defined. Here, we report a novel interaction between E. coli β-clamp and the helicase-nuclease RecBCD complex. Using bacterial two-hybrid assays and co-immunoprecipitation, supported by fluorescence microscopy, we show that RecB associates with β-clamp. Nuclear magnetic resonance spectroscopy maps the interaction to the canonical ligand pocket of β-clamp and identifies a clamp-binding motif in RecB (residues 1018–1023, QVEMEF), whose mutation abolishes binding. Functional assays indicate that this interaction occurs upon conformational switching of RecBCD at a Chi site, and disruption of the motif reduces survival after DNA damage. We also find indications of a second binding site in the helicase domain of RecB. These findings expand the β-clamp interactome and suggest a previously unappreciated role for β-clamp in DNA double-strand break repair, with potential implications for antibacterial strategies.
|
|
Scooped by
mhryu@live.com
Today, 1:24 AM
|
Different plasmids exist at different copy numbers per cell, and there is an inverse relationship between a plasmid's copy number and its size. Two recent studies quantified this relationship into a scaling law, but both the form and the interpretation of this law are contested. Here, I explore the issues with fitting a single law across plasmid diversity and suggest a consistent synthesis. First, I explore some potential problems with using sequencing-based estimates of copy number. Then, I discuss plasmid copy number through a series of case studies. I argue in favor of interpreting plasmid copy numbers not through a single law, but through the lens of two dominant evolutionary strategies. I suggest that small plasmids which lack active segregation mechanisms have a resulting tradeoff between plasmid inheritance and fitness cost to the host, which is responsible for an inverse relationship between copy number and size. In contrast, larger plasmids with active segregation mechanisms show a much weaker relationship, in line with evidence that their metabolic costs are dominated by the expression of specific genes rather than their size. Where plasmids in the 20-100kb range have higher copy numbers, I argue these probably arise more from selection at the level of the host cell for plasmid-associated phenotypes (e.g. antibiotic resistance) rather than from plasmid-level selection for inheritance.
|
|
Scooped by
mhryu@live.com
Today, 1:13 AM
|
The model plant Arabidopsis thaliana hosts diverse microbial communities collectively known as the microbiota. The plant microbiota is generally taxonomically structured and, in many cases, confers benefits to the plant host including plant growth promotion and enhanced stress tolerance. However, microbial imbalance can also result in deleterious effects, a phenomenon termed dysbiosis that was first coined in the gut microbiome field. To uncover the regulatory mechanism maintaining healthy plant homeostatic interactions with microbiota, we conducted screening using defined synthetic bacterial communities. We identified an Arabidopsis mutant displaying altered microbial profiles with an overall increase of microbial load and microbiota-dependent growth defects. Transcriptomic analyses combined with phytohormone quantification revealed that these phenotypes are attributed to an upregulation of the jasmonic acid (JA) signaling pathway in this mutant upon microbiota colonization. Furthermore, chemical treatment with different JA inducers reproduced similar phenotypes in wild-type plants, suggesting regulation through a positive feedback loop. Although activation of the JA pathway is typically associated with enhanced plant stress responses, our mutant exhibited reduced pathogen load at the expense of reduced plant growth and impaired salt tolerance. Together, our findings demonstrate that JA signaling not only orchestrates plant growth and defense but also plays a pivotal role in shaping plant–microbiota interactions. Controlled regulation of the JA signaling pathway is therefore essential to maintain balanced plant response to multiple environmental stressors.
|
|
Scooped by
mhryu@live.com
Today, 1:06 AM
|
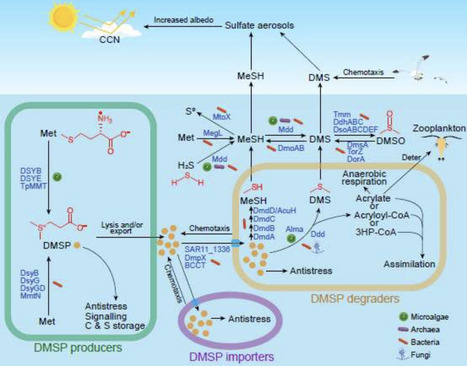
Ubiquitous marine microalgae and bacteria produce the abundant organosulfur compound dimethylsulfoniopropionate (DMSP) and/or catabolize it to climate-active gases, such as dimethylsulfide (DMS), with major consequences for global biogeochemistry and climate. However, their relative and dynamic roles in DMSP synthesis and catabolism remain poorly resolved, particularly during natural bloom events. Here, we combined metagenomics and metatranscriptomics, with measurements of intracellular/particulate DMSP (DMSPp), DMS concentrations and DMSPp production rates, as well as microscopy and flow cytometry, to predict the key microbes and enzymes driving DMSP/DMS dynamics during a spring–summer bloom in the Western English Channel. Microalgae and bacteria expressing the DMSP synthesis genes DSYB/DSYE and dsyB were likely major and significant DMSP producers, respectively, except during the largest observed DMSP spike. This spike coincided with elevated Synechococcus and autotrophic flagellate biomass but minimal DMSP synthesis gene expression. Axenic Synechococcus strains contained no detectable DMSP, implying flagellates with novel DMSP synthesis genes were likely responsible. Microbial DMSP import potential far exceeded catabolism, suggesting strong selection for DMSP uptake. Bacteria were the major predicted DMSP degraders, with DMSP demethylation potential dwarfing cleavage. However, the highest DMS concentrations were linked to Haptophyta expressing the DMSP lyase gene Alma, implying the significance of algal DMSP cleavage. Methanethiol-dependent DMS production was also likely important, with bacterial mddH transcripts coinciding with another major DMS spike. Overall, these results imply dynamic and contrasting roles of microalgae and bacteria, and their pathways, in coastal DMSP/DMS and sulfur cycling.
|
|
Scooped by
mhryu@live.com
Today, 12:57 AM
|
It is widely recognised that there is a significant gap between bacterial natural product potential and detected/described products. As such, there are several recent reviews on elicitation strategies for natural products discovery, which are often laboratory focused. Recently, a move to more ecology-focused approaches to understand the function of metabolites in nature and what impacts expression has been a growing trend. In this review, we aim to capture work done that goes beyond laboratory conditions and address ecological studies focussed on what impacts bacterial chemistry, covering both abiotic and biotic influences. We aim to touch on the impact of biodiversity loss, changes in ecosystems and future climate parameters and the implications this will have on natural products chemistry and biodiscovery efforts, the scale and consequences of which are not known.
|
|
Scooped by
mhryu@live.com
Today, 12:50 AM
|
Soil sulfur cycling plays a central role in ecosystem functioning and is tightly coupled to carbon–nitrogen–phosphorus–metal cycling. Key transformations occur at interfaces where microbes, minerals, and organic matter interact, yet these processes remain insufficiently resolved across scales and systems. Here, we synthesize evidence from multi-omics, isotopic tracing, and nanoscale spectroscopy and imaging to develop a microbial–mineral–organic matter interaction framework linking redox microdynamics, mineral reactivity, organic matter chemistry, and microbial guilds to sulfur speciation and turnover. We show how anthropogenic disturbance and climate change reshape interface-centered sulfur biogeochemical networks, with implications for nutrient retention, pollutant transport, and greenhouse gas emissions. We further identify major research hotspots, examine boundary conditions, and outline an artificial intelligence–process hybrid strategy for mechanism-based prediction and sustainable soil management under accelerating global change. This framework helps connect interface-scale mechanisms with ecosystem-scale consequences and provides a basis for future cross-system testing. Across soil systems, sulfur cycling is shaped by the interplay among microbial functional guilds, mineral reactivity, organic sulfur chemistry, and fluctuating physicochemical conditions, based on a framework of microbial–mineral–organic matter interactions.
|
|
Scooped by
mhryu@live.com
June 11, 4:43 PM
|
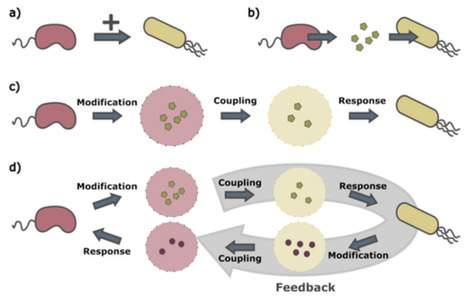
Microbial communities contribute to numerous processes that profoundly impact planetary and human health. They therefore hold potential for addressing many of today's pressing global challenges. Microbial ecosystems have been studied at many levels, ranging from the molecular processes of individual cells to the emergent properties and functions of the entire collective. One notable complexity of these ecosystems is that microbes are constantly engaged in interactions with their environment and other microbes, which in turn influences not only their own growth but also community function, assembly, and stability. While interactions are very often the subject of contemporary microbiology research, these studies often lack precise, mechanistically rooted characterizations of these interactions. In this article, we propose strategies to overcome such limitations by providing a conceptual framework for describing microbe–microbe interactions and discussing the implications of this framework for the study of microbial communities and their evolution. Starting from basic principles, we build a mechanistic description of microbial interactions that treats each interaction as a series of modular, interconnected subprocesses. We then examine how this modularity shapes microbial communities and their evolution, as well as how this modularity can improve our approaches for characterizing and mathematically modeling microbial ecosystems.
|
|
Scooped by
mhryu@live.com
June 11, 4:30 PM
|
Global surveys of microbial communities across biomes have shown that environmental variables such as depth and pH are strong determinants of community composition. However, we do not understand how the traits of individual taxa, and their evolutionary conservation, conspire to give rise to these patterns. Exploiting large-scale surveys of top soil and marine microbiomes, we use canonical correlation analysis (CCA) to concurrently infer directions of environmental variation and the associated compositional changes. We find that the primary canonical direction, capturing the dominant environmental gradient, exhibits a strong phylogenetic signal: individual species' responses to environmental shifts along this direction are similar among taxa with shared evolutionary history. In contrast, secondary canonical directions show weak or no phylogenetic structure. Together, these results suggest a two-scale view of microbial community assembly. Deeply evolutionarily conserved traits govern community reorganization along the main environmental driver of community composition. Additional environmentally driven changes in community composition then reflect traits that are more evolutionarily labile.
|
|
Scooped by
mhryu@live.com
June 11, 4:18 PM
|
Bilirubin, the predominant product of heme catabolism in mammals, enters the intestine via the hepatobiliary system and subsequently is metabolized by the gut microbiome. This process consumes bilirubin and generates multiple downstream derivatives, such as urobilinogen and stercobilinogen. Levels of bilirubin and its derivatives are associated with susceptibility to inflammatory and metabolic disorders, but the microbial species and enzymes that metabolize bilirubin have remained largely unknown. Here, demonstrate that metabolism of bilirubin to urobilinogen requires two separate reactions that can occur in either order and identify novel enzymes and pathway intermediates required for conversion. We find that bilirubin reductase (BilR), an enzyme that was recently discovered and proposed to convert bilirubin to urobilinogen, is specific for reducing the methine bridges of bilinoids, converting bilirubin to the novel intermediate divinylurobilinogen and mesobilirubin to urobilinogen. Using transcriptomic profiling, we identify the bilinoid vinyl reductase (BilV) responsible for reducing the vinyl groups of bilirubin and divinylurobilinogen. BilV is a flavin-dependent oxidoreductase of the Old Yellow Enzyme (OYE) superfamily with a broad distribution across human gut bacteria that overlaps with but does not completely mirror the distribution of BilR. These findings establish the complete pathway for bacterial conversion of bilirubin to urobilinogen, enabling defined studies to interrogate how this metabolism contributes to human health and disease.
|
|
Scooped by
mhryu@live.com
June 11, 4:11 PM
|
Commercially available make-on-demand libraries now exceed 100 billion compounds, requiring over 50 years to screen on 2,000 CPU cores using conventional docking. We present two complementary approaches to address this challenge. CombiDOCK, a combinatorial docking framework, enables exhaustive screening at the 100-billion scale within 40 days. MINT-Dock, a generative framework, accelerates navigation of this space by integrating CombiDOCK with Monte Carlo Tree Search. Benchmarked on 46 diverse targets, CombiDOCK matched full-molecule docking accuracy, and MINT-Dock achieved a 4,800-fold enrichment over random selection. Compared with prior billion-scale brute-force campaigns against σ2, VMAT2, and VAChT, prospective CombiDOCK screens of the 100-billion-molecule library yielded higher hit rates and more potent ligands, while MINT-Dock achieved comparable outcomes across single- and multi-target objectives with >20-fold computational cost reductions. Docking-predicted poses of the best VAChT-binding compounds were confirmed by cryo-EM structures. These methods provide exhaustive and generative paths for navigating the trillion-molecule frontier of drug discovery.
|
|
Scooped by
mhryu@live.com
June 11, 3:58 PM
|
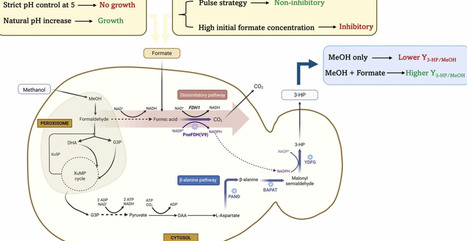
Komagataella phaffii is a promising cell factory that can use CO2 derived methanol, a sustainable carbon (C1) source, for chemical production. Introducing the ß-alanine biosynthetic pathway alongside a NADP+-dependent formate dehydrogenase (FDH) in K. phaffii enables the production of the platform chemical 3-hydroxypropionic acid (3-HP). Co-feeding of formate and methanol (MeOH) was systematically explored to enhance cellular reducing power and improve 3-HP biosynthesis. Implementing a pulsed formate strategy alongside MeOH resulted in up to a 20.7% increase in 3-HP per gram of MeOH (Yg3-HP gMeOH-1) compared to MeOH alone in shake flask cultivations. This co-feeding strategy likely enhanced NADPH availability through formate oxidation via the introduced NADP⁺-dependent FDH, thereby improving redox balance, as supported by simulation studies based on the K. phaffii genome-scale metabolic model. Similar improvements were demonstrated in repeated batch cultivations at 1-L bioreactor scale, where a formate pulse every 4-hour led to a 25.8% increase in Yg3-HP gMeOH-1 and a 41% increase in volumetric productivity over the control without formate (only MeOH). In addition to the feeding strategy, pH regulation also played a crucial mechanistic role: strict pH control at 5 inhibited growth, due to the predominance of undissociated formic acid, whereas allowing the pH to rise to 6-7 favored dissociation and supported higher productivity. These findings elucidate the pH dependent nature of formate assimilation and highlight the potential of coupling MeOH and formate co-utilization with dynamic feeding and pH strategies to enhance bioproduction in K. phaffii.
|
|
|
Scooped by
mhryu@live.com
Today, 2:11 AM
|
Functional genomics have been hampered by the paucity of efficient methods that connect genotype and metabolic phenotype at single-cell resolution. Using the industrial microalga Nannochloropsis oceanica as a model, we introduced a platform that comprises a genome-wide single-gene-edited mutant library and high-throughput Raman-activated cell sorting (RACS). The CRISPR/Cas-generated library consisted of 3567 microalgal mutants derived from 2397 effective guide RNAs. Label-free sorting of the library for high carotenoid content by RACS unraveled mutations in the violaxanthin de-epoxidase (noVDE) or in the proteasome assembly chaperone 4 (noPAC4) genes. Knocking out all five known noVDEs revealed that the high carotenoid content is due to violaxanthin increase, whilst noPAC4 knockout boosted carotenoid content with elevations in violaxanthin, zeaxanthin, and β-carotene. Genetic and transcriptomic evidence suggested two previously unknown modes of carotenogenesis regulation mediated by noPAC4: epigenetic mechanisms via histone deacetylase (HDAC) and post-translational controls by the 26S proteasome. Therefore, by label-freely sorting single-cell metabolic phenotype and rapidly yet unambiguously tracing it to a genotype, this forward-genetics approach can greatly accelerate the discovery of genes and pathways. Functional genomics in algae are limited by efficient methods that connect genotype and metabolic phenotype at single-cell resolution. Here the authors introduce an approach based on a genome-wide mutant library and a Raman Cytometer, discovering a carotenoid synthesis regulating pathway.
|
|
Scooped by
mhryu@live.com
Today, 2:06 AM
|
The precise translational control of gene expression by small molecules through RNA-based switches holds considerable promise for both research and therapeutic developments. However, current high-performance RNA switches remain limited in adaptability, with strong responses typically constrained to a narrow set of specific ligand-aptamer pairs. To address this limitation, we introduce a robust and generalizable RNA platform based on a multivalent aptamer design, which significantly enhances ligand-responsive protein expression through alternative splicing regulation. We have demonstrated that the inherently weak aptamers, such as those for theophylline or tetracycline, can be dramatically improved through this multivalency circuit, elevating the induction levels from modest (<10-fold) to over 100-fold, an increase of more than an order of magnitude. Leveraging these improved switches, we achieve multiplex and orthogonal control over distinct protein outputs with these suboptimal aptamers. Furthermore, we implement precise manipulation of cellular phenotypes through the ligand-controlled expression of functional proteins, including the pro-apoptotic effector BAX and the adhesion protein E-cadherin. This work establishes a general and adaptable RNA platform for expanding the toolbox of small-molecule regulators of protein expression, with potential applications across synthetic biology and therapeutic applications.
|
|
Scooped by
mhryu@live.com
Today, 1:33 AM
|
RNA devices, including riboswitches, aptazymes, and RNA-based biosensors, have become essential components in synthetic and molecular biology. These systems make use of RNA’s modularity and structural diversity to build programmable tools for sensing, regulation, and cellular computation. To drive these systems, RNA aptamers usually need to undergo a conformational change upon ligand binding. However, the limited availability of such aptamers has restricted the development and application of RNA devices, particularly in mammalian systems. Traditional selection methods often prioritize high-affinity binding, resulting in a scarcity of conformation-switching aptamers. Here, we addressed this limitation by selecting a caffeine-binding aptamer with RNA Capture-SELEX and in vivo screenings. This aptamer functioned as a modular regulator of riboswitches and ribozymes in Saccharomyces cerevisiae and mammalian cells. Through new optofluidic screenings, we overcame throughput and sequence-function challenges inherent in ribozyme screening for mammalia. Additionally, a straightforward grafting method transformed the aptamer into a fluorogenic iSpinach aptasensor. This work demonstrates the successful selection of a modular and communicating aptamer for a challenging target like caffeine and establishes robust strategies for their modular integration into diverse RNA platforms. These strategies pave the way for broadening the repertoire of aptamers and expanding the potential of RNA-based synthetic biology.
|
|
Scooped by
mhryu@live.com
Today, 1:18 AM
|
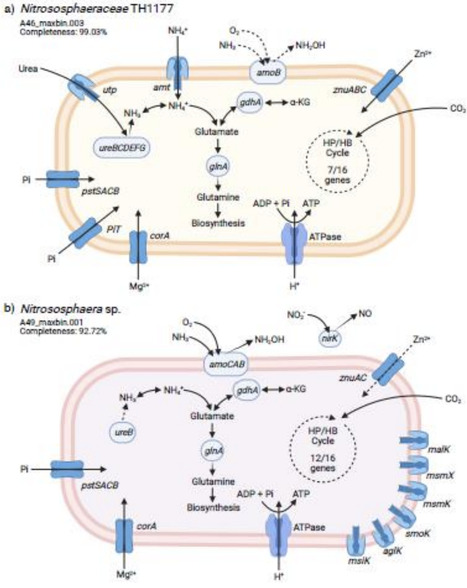
The soil matrix is a heterogeneous mixture composed of aggregates – three dimensional complexes composed of organic materials and mineral particles. Soil aggregates vary considerably in physical and chemical properties by size, making them unique habitats for distinct microbial communities and metabolic pathways. Yet, this microscale spatial variability is often overlooked in studies that use homogenized soil cores. We investigated the microbial taxonomy, functional gene composition, and metabolic products observed in four aggregate size fractions ranging from 8 mm to free particles (below 53 μm) collected from agricultural soils under two different management practices. The functional gene composition differed significantly among aggregate sizes, with higher abundances of genes for the degradation of plant-derived compounds in the macroaggregates and for biomass recycling in the two smallest size fractions. These differences corroborated with significant differences in the composition of the metabolome but not specific enzyme activities. Both taxonomic profiling and reconstruction of genomes from metagenomes revealed a higher abundance of ammonia-oxidizing archaea in the macroaggregates in comparison to other aggregate sizes, and analysis of their genomes revealed complementary metabolisms potentially enabling them to colonize different niches within the same habitat. Together, our results show that soil microbial communities and their functions are shaped by the size of soil aggregates, likely driven by differences in resource availability between macro- and microaggregates.
|
|
Scooped by
mhryu@live.com
Today, 1:09 AM
|
Microbial communities are often more species-rich than predicted from classical ecological models. The high levels of coexistence observed in nature are typically attributed to forces that modulate niche availability and stabilize communities. Specific drivers of niche partitioning are often tested in isolation, and the interactive effects of niche variation across resources, space, and time have not been tested together experimentally to determine how they affect community responses. Here, we used 26 bacterial strains previously isolated from carnivorous pitcher plant (Sarracenia purpurea) aquatic pools to construct and expose species-rich synthetic communities to four factors that alter environmental complexity in a fully factorial design, creating combinations of resource complexity, spatial niche structure, and temporal fluctuations. Across treatments, increased niche complexity generally, but not always, promoted the long-term retention of more species, with a saturating effect at the highest levels of complexity. Resource complexity emerged as a primary driver of diversity, with its effects also depending on other niche axes. Interactions among factors frequently deviated from additive expectations, with both synergistic and antagonistic effects observed depending on the combination of conditions. Together, these results show that environmental complexity shapes bacterial diversity through context-dependent, nonlinear interactions among niche dimensions, highlighting that the relationship between niche dimensionality and diversity is contingent on how environmental factors combine.
|
|
Scooped by
mhryu@live.com
Today, 1:00 AM
|
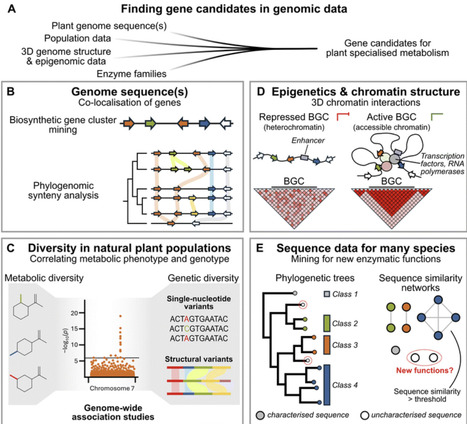
Some of the most prominent natural products originate from plants. Discovering their biosynthetic genes has been a slow process. In contrast to microbial systems, co-expression analysis rather than genome mining has been the main strategy to elucidate biosynthetic pathways in plants. However, traditional co-expression analyses are limited in efficiency and often not as successful as desired. In this review, we describe emerging technologies to improve or replace traditional co-expression analyses, for example based on genome or protein data. Furthermore, we critically discuss the current state and impact of artificial intelligence and machine learning in the field. Our review will help to select the most efficient approaches for elucidation of biosynthetic pathways in plants for future work. Additionally, we highlight areas that require further methodological improvements to guide future research.
|
|
Scooped by
mhryu@live.com
Today, 12:56 AM
|
Prokaryotic gene regulation is governed by the dynamic conformation of the transcription machinery and the nucleoid. To navigate environmental fluctuations, prokaryotes reprogram RNA polymerase (RNAP) via σ factors and transcription factors (TFs) that remodel DNA conformation. Bacteriophages, in turn, exploit these mechanisms to hijack host gene expression. This review dissects recent structural insights into transitions governing transcriptional regulation, highlighting that both TFs and nucleoid-associated proteins act as ‘architects’ to remodel DNA structure, thereby blurring the traditional distinction between site-specific regulators and global genome organizers. By linking RNAP and DNA remodeling to broader prokaryotic conflicts, from stress adaptation to arms races between hosts and mobile genetic elements, the authors underscore the profound biological impact of perturbing prokaryotic transcriptional architecture.
|
|
Scooped by
mhryu@live.com
Today, 12:41 AM
|
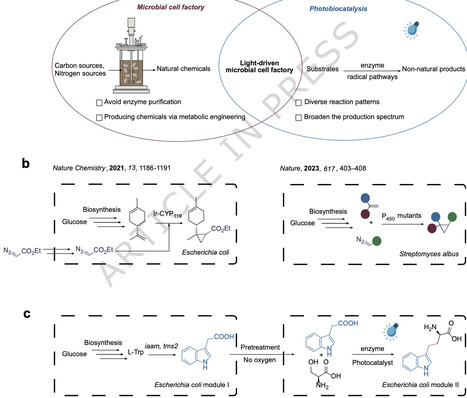
Microbial cell factory is a powerful biological tool for synthesizing value-added molecules due to its unmatched sustainability and selectivity. However, the inherent catalytic specificity of natural enzymes limits product diversity. Although photobiocatalysis has expanded enzyme catalytic capabilities, challenges such as laborious enzyme purification, incompatibility with in vivo conditions, and complex unnatural substrate synthesis hinder photobiomanufacturing efforts. Herein, we combine a microbial cell factory with a new-to-nature photobiocatalytic transformation via a modular design that combines aerobic fermentation (Module I) and anaerobic photocatalysis (Module II) to achieve de novo biosynthesis of d-homotryptophan. In Module I, we engineer E. coli equipped with a biosynthetic gene cluster to produce indole-3-acetic acid (IAA) via modifying metabolic flux and multi-copy genetic amplification strategies. In Module II, we develop an efficient synergistic photoredox/enzymatic synthesis of d-homotryptophan from l-serine and IAA by a pyridoxal phosphate (PLP)-dependent tryptophan synthase beta-subunit (TrpB) variant. This work synergistically merges emerging photobiocatalytic reactivity with natural biosynthesis, demonstrating a platform with potential for future biomanufacturing of non-natural products. Microbial cell factories are a powerful tool for synthesizing value-added molecules, but the inherent catalytic specificity of natural enzymes limits product diversity. Here, the authors combine a microbial cell factory with a new-to-nature photobiocatalytic transformation via a modular design that combines aerobic fermentation and anaerobic photocatalysis to achieve de novo biosynthesis of d-homotryptophan.
|
|
Scooped by
mhryu@live.com
June 11, 4:35 PM
|
Correlations between cellular variables, such as gene-expression levels, provide insights into regulatory mechanisms. We focus here on correlations between mRNA and protein levels and re-examine previously derived analytical predictions. We test this prediction on single-cell E. coli data and see substantial disagreement. We hypothesize that this discrepancy arises from the assumption of constant cell volume and develop a theoretical framework for mRNA–protein correlations in growing and dividing cells. Within this framework, we derive an analytical expression for mRNA– protein correlations and show that explicit incorporation of growth and division substantially alters these correlations. The resulting relation is invariant to upstream transcriptional dynamics, and we validate it using stochastic simulations across multiple gene-regulatory architectures. Finally, we show that the derived predictions are consistent with the E. coli data.
|
|
Scooped by
mhryu@live.com
June 11, 4:27 PM
|
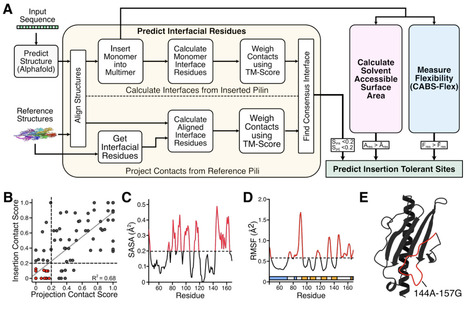
Bacterial surface structures have enabled display systems with broad impact across biotechnology, but their narrow host range limits their deployment into diverse species. Conversely, Type IV Pili (TFP, T4SS) are ubiquitous, structurally conserved appendages found across bacteria, but have been minimally explored for display. Here, we describe a computational framework for predicting viable insertion sites in major pilins for stable TFP-mediated display, which we apply to the major pilin PilA1 of the cyanobacterium Synechocystis sp. PCC 6803 to enable covalent binding to living materials. By analyzing an Alphafold3-generated PilA1 monomer alongside known multimeric TFP multimers, our pipeline identifies non-interfacial, solvent-accessible, and flexible sites for optimal PilA1 display. We probe these sites with both full-length and truncated SpyCatcher003 at two different expression levels. We show that cells expressing these PilA1-SpyCatcher 003 fusions maintain up to 8-fold higher levels of cell suspension than previous C-terminal PilA1 display platforms, suggesting improved TFP assembly despite more than a two-fold increase in cargo size. Additionally, we validate SpyCatcher003 reactivity across the engineered strains, enabling covalent attachment of SpyTag 003-containing proteins on the Synechocystis surface. Lastly, we utilize this covalent patterning to achieve a four-fold increase in Synechocystis loading into a living material without compromising its viscoelastic or mechanical properties. Taken together, this work provides a predictive framework for TFP engineering, and opens the door towards programmable surface display across the breadth of bacterial species.
|
|
Scooped by
mhryu@live.com
June 11, 4:14 PM
|
Spurious protein sequences, resulting from gene prediction errors, theoretically should not yield folded structures. AlphaFold2 was previously shown to predict short spurious sequences with high pLDDT scores and was therefore unlikely to distinguish between real proteins and spurious proteins which are usually short. We evaluate whether newer structure prediction methods (ESMFold and AlphaFold3) similarly predict short sequences with high pLDDT or if they better discriminate between spurious and real proteins. All three structure prediction methods (ESMFold, AlphaFold2, and AlphaFold3) predict short spurious sequences from AntiFam with unexpectedly high pLDDT scores, however the discrimination between spurious and real proteins improves beyond 100 amino acids. By analysing sequences with disparate pTM and pLDDT scores, we identified two likely spurious shadow ORFs in Swiss-Prot and one potentially non-spurious AntiFam entry. Using the structure prediction scores, we developed a Gaussian Process Model and evaluated its performance on AlphaFold DB, identifying potential spurious proteins at scale. While limited on its own, this model can increase confidence in spurious protein identification when combined with other methods.
|
|
Scooped by
mhryu@live.com
June 11, 4:03 PM
|
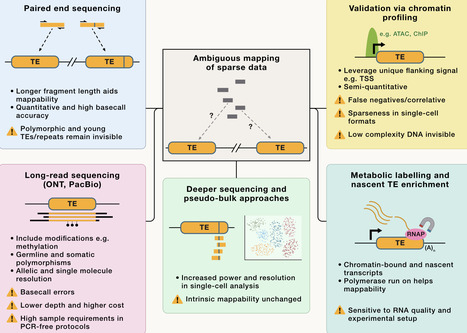
Transposable elements (TEs) comprise nearly half of mammalian genomes and have shaped genome architecture, chromatin organization, and transcriptional landscapes. Thanks to recent advances in long-read sequencing and functional (epi)genomics, the focus has shifted from TE families to individual TE loci, revealing widespread, locus-specific regulatory roles. While most TEs have lost the capacity to mobilize, they still retain a DNA form and, when transcribed, an RNA form, both of which can affect genome regulation. TEs can serve as alternative promoters, exons, splicing regulators, and 3′ end modulators. They can also act as enhancers, drive three-dimensional (3D) genome organization, and give rise to long non-coding RNAs (lncRNAs) that serve as platforms for transcriptional and chromatin regulators. Mechanistically, TE repression involves DNA methylation, histone modification, phase-separated condensates, RNA modifications, RNA degradation, and nuclear compartmentalization, yet this repression can be selectively lifted during development or stress to expand regulatory potential. TEs therefore contribute to cell-type identity, developmental transitions, and responses to environmental stimuli, while their dysregulation is linked to human disorders including neurodegeneration, cancer, and autoimmune disease. TEs also hold translational promise as biomarkers and tools for gene and cell engineering. In summary, the pervasive integration of TEs as mini-genes, structural scaffolds, and regulatory elements redefines our view of the genome: rather than a gene-centric landscape dotted with repetitive “junk,” mammalian DNA is a TE-rich ecosystem in which TEs drive gene regulatory networks and evolution.
|
























r-2st. Necromass