 Your new post is loading...

|
Scooped by
mhryu@live.com
Today, 4:20 PM
|
Peptides, as therapeutic molecules, offer unique advantages in targeting complex protein surfaces, yet their rational design remains limited by the vastness of the sequence space and the constraints of traditional approaches. Here, we propose High-PepBinder, a sequence-only conditional diffusion framework for target-specific peptide generation. Guided by the target protein sequence, High-PepBinder adopts a dual encoder architecture that integrates protein language models (pLMs) with the diffusion model. This approach cascades the peptide generation model with an affinity classifier and enables the generation process to capture affinity-related features of the peptides through lightweight joint optimization. Due to the scarcity of protein-peptide affinity data, we constructed PepPBA, to our knowledge the most comprehensive dataset to date, and established a structure- and physics-based screening pipeline to prioritize top candidates. Results show that High-PepBinder demonstrates competitive performance across multiple peptide generation and affinity-related tasks. For representative targets, including KEAP1, XIAP, and EGFR, the generated peptides preserve key binding geometries and interface patterns of reference peptides in predicted complexes, while maintaining sequence diversity and favorable predicted properties. Overall, High-PepBinder contributes toward a general and sequence-only strategy for peptide design, offering a computational framework for expanding peptide discovery against challenging targets.
|
|
Scooped by
mhryu@live.com
Today, 4:02 PM
|
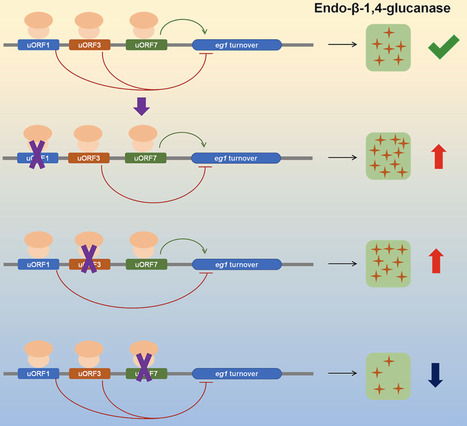
Cellulase plays an irreplaceable role in biomanufacturing using plant biomass as feedstock. However, improving cellulase production by fungi through manipulation of upstream open reading frames (uORFs) in the 5′-untranslated regions (5′-UTR) of cellulase genes has been less frequently explored. This study aimed to screen uORFs in the 5′-UTR of cellulase genes in Penicillium oxalicum, identify functional uORFs in the 5′-UTR of the eg1 gene which encodes a key endo-β-1,4-glucanase (EG) in P. oxalicum, and enhance fungal cellulase production through uORF modifications. Among the 25 cellulase genes examined in P. oxalicum strain HP7-1, 23 contained uORFs in their 5′-UTR. Seven uORFs were annotated in the 5′-UTR of the eg1 gene. A uORF-green fluorescent protein (GFP) reporter system demonstrated that uORF1 and uORF3 inhibited, while uORF7 enhances, GFP abundance. Overexpression of eg1 containing uORF1 or uORF3 variants where the start codon of the uORF was mutated to AAG in P. oxalicum led to a significant 91.7 % and 62.1 % average increase in carboxymethyl cellulase production after 4 days of induction compared to the start strain ΔPoxKu70. Real-time quantitative reverse transcription-polymerase chain reaction, mRNA stability determination, and in vitro translation experiments collectively revealed that these three uORFs influence the mRNA stability of the downstream mORF, but not translation efficiency. These findings highlight the critical role of uORFs in regulating gene expression during fungal enzyme biosynthesis and offer a valuable alternative strategy for improving enzyme production.
|
|
Scooped by
mhryu@live.com
Today, 3:51 PM
|
Tailoring natural enzymes to synthetic needs is often associated with high costs and long timelines, hindering the broader adoption of biocatalysis in the chemical and pharmaceutical industries. To address this, we developed the RISE (rapid in vitro semi-rational engineering) workflow that makes enzyme engineering accessible to chemistry laboratories. RISE integrates four key concepts: computational design of focused variant libraries, rapid generation of linear mutant DNA libraries via PCR, cell-free protein synthesis from linear template DNA, and iterative cycles of mutagenesis, expression, and testing to accumulate beneficial mutations. In a proof-of-concept study, we engineered a ketimine reductase from Rattus norvegicus (RnKIRED), achieving stereoselectivity inversion in one reductive amination reaction and a 400-fold activity improvement in another. These engineered variants enabled the gram-scale synthesis of key intermediates for ACE2 inhibitor drugs. RISE bridges the gap between inefficient wild-type enzymes and expensive directed evolution, promoting biocatalysis implementation in early chemical development.
|
|
Scooped by
mhryu@live.com
Today, 3:42 PM
|
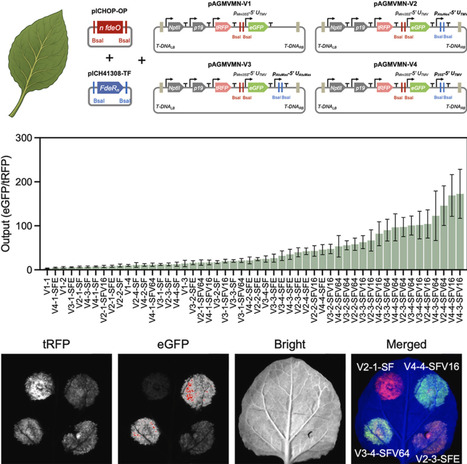
Advancing plant synthetic biology requires an abundant supply of orthogonal and tunable genetic parts to express multiple genes simultaneously. Current genetic parts, particularly promoters, used in plants are still limited and often suffer from tissue specificity and endogenous regulation in planta. Synthetic promoter systems that combine engineered plant-compatible transcription factors (TFs) with synthetic promoters provide a promising alternative approach to enrich current plant synthetic biology toolkits. Leveraging the feature that LysR-type TFs usually bind to the operators regardless of the presence or absence of ligands, we present a systematic approach to develop and characterize a large suite of synthetic promoter systems based on a single LysR-type bacterial TF. Using FdeR from the soil bacterium Herbaspirillum seropedicae and its corresponding operator, fdeO, we developed 52 synthetic promoter systems regulated by four FdeR-derived synthetic activators. Transient expression assays in Nicotiana benthamiana showed that the synthetic promoters were constitutively activated by synthetic activators in a ligand-independent manner, and the resulting promoters spanned a wide range of expression activities, with the lowest equivalent to minimal CaMV 35S promoter and the highest with ∼65% of CaMV 35S activity. We also developed modular workflows and pipelines to accelerate the development of the synthetic transcription platform, including a MoClo-based plug-and-play assembly system for plant-compatible promoter construction and a yeast-based prescreening platform for rapid TF evaluation in eukaryotic cells. This work has demonstrated a scalable framework for developing a large set of synthetic promoter systems from minimal genetic components, leveraging bacterial TF diversity to expand the plant synthetic biology toolkit for robust, orthogonal gene expression.
|
|
Scooped by
mhryu@live.com
Today, 3:31 PM
|
Poor aqueous solubility of steroid precursors, such as pregnenolone and progesterone, limits microbial biotransformation and high-throughput strain screening, representing a bottleneck for strain improvement and potential industrial applications. To address this, we developed a growth-coupled progesterone-responsive biosensor in Saccharomyces cerevisiae, integrated with a hydroxypropyl-β-cyclodextrin (HP-β-CD) system to enhance intracellular steroid availability. The biosensor links progesterone formation to cell growth and fluorescence, with selection stringency finely tuned via an IPTG-inducible lac operator and 3-aminotriazole (3-AT) to suppress low-producing cells. Coupled with atmospheric and room temperature plasma (ARTP) mutagenesis, the growth-coupled biosensor–FADS platform identified five yeast variants capable of improved conversion of pregnenolone to progesterone while expressing 3β-hydroxysteroid dehydrogenase (3β-HSD) without altering the enzyme itself. The progesterone production of these selected variants was subsequently validated using 1 mM pregnenolone as the substrate, showing 2.0–3.37-fold higher titers than the wild-type strain, demonstrating proof-of-concept. Microfluidic droplet encapsulation allowed clear separation of high-producers, highlighting the platform's selectivity, robustness, and scalability. This synthetic biology–driven system integration platform provides a practical, modular, and high-throughput strategy for screening poorly water-soluble steroid-producing yeast. It is adaptable to other bioactive molecules, can support future enzyme evolution, and demonstrates potential for broader biotechnological applications.
|
|
Scooped by
mhryu@live.com
Today, 12:43 PM
|
Invasive pest Spodoptera frugiperda, known as the fall armyworm (FAW), evolves rapid resistance to chlorantraniliprole (CAP) via symbionts, reducing traditional control efficacy and causing economic losses. To address the formidable challenge of insecticide resistance, we introduce phage therapy into pest control, enabling precise targeting and efficient lysis of symbionts that mediate resistance. We employ zein to synchronously encapsulate phages and insecticides, constructing a nano-insecticide. This nano-insecticide ensures stability, exhibits robust performance by protecting phages against temperatures up to 60°C, and enhances their survival under UV irradiation by 83-fold. It intelligently responds to the pest gut enzymes for precise and controlled release, improving FAW control by 17% and overcoming resistance. Additionally, pesticide residue is reduced by 82.4%, with minimal impact on soil and maize microbial communities, preserving seedling growth. This modular, eco-friendly framework offers a sustainable solution for resistant pests, addressing the escalating challenge of resistant pests and paving the way for advancements in sustainable agriculture.
|
|
Scooped by
mhryu@live.com
Today, 12:09 PM
|
The biotransformation of acetaminophen, a pharmaceutical contaminant widely found in crop produce, in the rice phyllosphere highlights critical xenobiotic-plant-microbiota interactions. This study investigated acetaminophen uptake, translocation, and transformation in hydroponically exposed rice shoots, revealing accumulation at 8.33 ± 0.82 μg/g and conversion into hydroxylated, glycosylated, methylated, thiomethylated, sulfonated, dimerized, and amino acid-conjugated derivatives. Specifically, these transformations may be driven by plant enzymes (cytochrome P450, glycosyltransferases, sulfotransferases, and methyltransferases) and synergistically by enriched microbial genera (Sphingomonas, Pantoea, and Pseudomonas). Furthermore, acetaminophen stress altered the rice phyllosphere metabolome (elevated linoleic acid and jasmonic acid) and reshaped microbial communities, with enhanced degradation pathways and network complexity indicating adaptive stress mitigation. Overall, this integrated transcriptome, metabolome, and microbiome profiling provides mechanistic insights into the cooperative detoxification role of plant enzymes and phyllosphere microbes, offering perspectives on leveraging plant-microbiota interactions to reduce xenobiotic impacts on crops and food safety.
|
|
Scooped by
mhryu@live.com
Today, 11:51 AM
|
Adaptive, closed-loop control of cellular behavior is essential for next-generation therapies, yet most current treatments operate in an open-loop manner and lack robustness to patient variability and disease dynamics. Here, we establish a control-theoretic platform for rational engineering of closed-loop cell-based therapies that achieve precise and robust regulation. First, we introduce multi-dimensional nullgram profiling, a high-throughput approach that enables quantitative prediction and design of advanced genetic controllers in human cells across circuit topologies and parameter regimes in a single experiment. To evaluate dynamic therapeutic behavior, we next develop Cyberpatient-in-the-loop, an optogenetic digital twin platform that interfaces engineered mammalian cells with computational disease models, enabling systematic testing of closed-loop performance under realistic perturbations. Finally, we leverage these approaches to implement integral feedback cell therapies that sense inflammatory signals and autonomously regulate cytokine levels in primary immune cell cultures. Together, these results establish a general paradigm for engineering cell-based control systems and provide a foundation for next-generation cell therapies.
|
|
Scooped by
mhryu@live.com
Today, 10:32 AM
|
Large language models (LLMs) have shown remarkable success in natural language processing, prompting interest in their application to genomic sequence analysis. Genomic Language Models based on similar architectures offer a promising avenue for synthetic genome generation and characterization. However, their effectiveness for biological sequence modeling remains poorly characterized. We present a comprehensive evaluation of a state-of-the-art genomic Language Model (gLM), Evo 2, on multiple genomic reconstruction tasks. We tested Evo 2 on diverse prokaryotic, eukaryotic and viral genomes and assessed performance across key biological features and organizational patterns. Our results reveal systematic failures in gLM-based genomic reconstruction. While the synthetic sequences captured local sequence statistics, they consistently failed to preserve long-range genomic organization, repeat and k-mer composition, transcription factor binding site architecture, and evolutionary constraints. Generated sequences exhibited severe violations of natural genomic patterns and models showed particular difficulty with repetitive elements. These evaluation criteria provide a set of biologically grounded benchmarks for assessing the quality and realism of synthetic genomes. These findings suggest fundamental limitations in current gLM architectures for capturing the hierarchical, evolutionarily-constrained nature of genomic sequences. Our work highlights the need for specialized architectures that explicitly model biological constraints rather than relying solely on statistical patterns, with important implications for computational biology applications requiring realistic sequence generation.
|
|
Scooped by
mhryu@live.com
Today, 10:23 AM
|
Enhancing enzyme thermostability is crucial for industrial applications requiring robust performance under extreme conditions. Structure-based protein design models excel at improving thermal stability but often compromise enzymatic activity, while sequence-based models better preserve activity but struggle to enhance thermostability. It is challenging to efficiently generate multi-site mutants with both improved thermostability and intact activity using minimal experimental effort. Here, we used zearalenone hydrolase (RmZHD) and xylanase as model systems to evaluate different strategies for multi-site mutation design: (i) structure-based design with the ABACUS-R model, (ii) sequence-based design with the ProGen2 or MSA Transformer, (iii) integrated approaches combining either ProGen2 or MSA Transformer with ABACUS-R via Markov Chain Monte Carlo sampling with multi-objective scoring. Results showed that designing with ABACUS-R increased thermal stability by ~15°C but caused 80–100% activity loss. Sequence-based designs retained ~19% wild-type activity but failed to improve thermostability. Notably, zero-shot designs from integrating ABACUS-R with MSA Transformer achieved significant thermostability gains (ΔTm ~8°C) while preserving >95% wild-type activity. This highlights the potential of combining sequence-and structure-based deep learning models for developing industrially relevant thermostable enzymes.
|
|
Scooped by
mhryu@live.com
Today, 10:07 AM
|
Transcriptional regulation involves complex and dynamic protein–DNA interactions, which alter chromatin states and, consequently, regulate gene expression. In plants, current technologies face challenges in efficiently capturing dynamically DNA-binding proteins, especially transcription factors. Here, by leveraging the binding ability of dCas9 to specific DNA fragments and the labelling capacity of the TurboID protein for adjacent proteins, we have developed a CRISPR-based sequence proximity binding protein labelling system (CSPL) to detect promoter-binding proteins. Using this approach, we identified both known and novel upstream binding proteins on the PIF4 promoter in Arabidopsis, cabbage and rice. This demonstrates the powerful capabilities and broad potential applications of CSPL for detecting promoter-binding proteins in plants. The authors developed a CRISPR-based proximity labelling system to profile DNA-binding proteins in plants, especially the transcription factors of target genes, as exemplified by studies on the regulators of PIF4 in Arabidopsis, cabbage and rice.
|
|
Scooped by
mhryu@live.com
January 18, 12:37 PM
|
Drought stress severely constrains crop productivity, and while plant growth-promoting rhizobacteria (PGPR) are known to enhance drought tolerance by modulating host aquaporins (AQPs), the specific role of bacterial biofilm formation in this regulatory process remains poorly understood. Here, we demonstrate that biofilm formation is a pivotal mechanism through which Bacillus velezensis D103 confers drought resilience to maize. Under drought stress, maize root exudates synergistically enhanced D103 biofilm formation, which was essential for robust root colonization and mediated a drought-adaptive restructuring of the rhizosphere microbiome. Crucially, we found that an intact bacterial biofilm systemically upregulated key plant AQPs (ZmPIP2;6 and ZmTIP1;1), thereby enhancing root water transport capacity. Using virus-induced gene silencing, we further clarified the molecular mechanism underlying this biofilm-aquaporin link, revealing that ZmPIP2;6 is indispensable for D103-conferred drought tolerance. Our findings refine the current understanding of PGPR-mediated drought tolerance, highlighting that biofilms coordinate host AQP expression, rhizosphere microbiome assembly, and soil water retention to enhance drought resilience. This work provides a mechanistic basis for developing effective microbial inoculants.
|
|
Scooped by
mhryu@live.com
January 18, 12:11 PM
|
Spatial engineering has emerged as a transformative paradigm for orchestrating metabolic flux through biomolecular compartmentalization. In cellular systems, the cytosolic dispersion of heterologous enzymes and evolutionary-driven metabolic priorities of native pathways necessitate spatial solutions that transcend conventional enzyme engineering. Concurrently, in vitro metabolons provide critical mechanistic insights into enzymatic cascade reactions through controlled assembly. This review systematically evaluates several spatial engineering platforms for biocatalytic process control—including scaffolded compartments (liposomes, DNA origami, polymersomes, and bacterial microcompartments) and scaffoldless assemblies (membraneless organelles and coacervates)—designed to reconfigure metabolic landscapes in cellular or cell-free contexts. Through critical analysis of recent advances in model construction and functionalized applications, we establish a framework for understanding different spatial control principles governing pathway efficiency and flux redistribution. Finally, we conclude with a comprehensive assessment of current limitations in mechanistic elucidation, dynamic regulation and cross-system compatibility, while projecting future developments towards multifunctional spatial organization tools and biomimetic platforms for synthetic biology and cellular engineering.
|
|
|
Scooped by
mhryu@live.com
Today, 4:14 PM
|
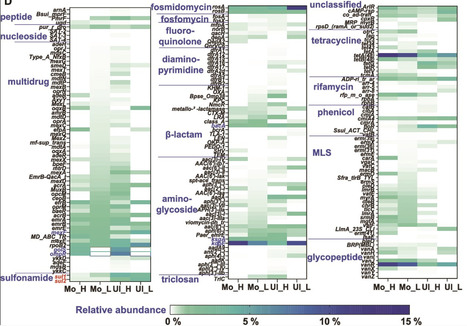
Soils act as both reservoirs and filters of antimicrobial resistance genes (ARGs); however, the ecological and genetic traits of antibiotic-degrading bacteria (ADB) and their interactions with nondegrading bacteria (NADB) across soil types remain poorly understood. In particular, the role of ADB in ARG dynamics and their potential contribution to horizontal gene transfer (HGT) are still underexplored. Here, we applied 13C-DNA stable isotope probing (DNA-SIP) combined with metagenomic sequencing to resolve active ADB from NADB in two contrasting soils: Ultisol and Mollisol. ADB harbored significantly more abundant and diverse chromosomal ARGs — especially multidrug and tetracycline resistance genes — often co-localized with mobile genetic elements (MGEs) and degradation genes, suggesting robust and regulated resistance strategies. In contrast, NADB relied more on plasmid-borne ARGs, reflecting flexible but potentially transient adaptation. Soil properties shaped both resistome composition and host taxa. Mollisol enriched enzymatic degraders such as Lysobacter and Nocardioides, while Ultisol favored stress-tolerant Burkholderia, which carried up to 34 ARGs and exhibited membrane-associated resistance. Notably, 89 ARGs or MGEs were found co-localized with degradation genes on assembled contigs, highlighting a strong potential for HGT. In addition, 24 high-potential ARG hosts were identified, including Ralstonia pickettii and Saccharomonospora viridis. These findings reveal that antibiotic degradation is embedded within complex, soil-specific resistome networks. This work enhances our understanding of ARG ecology and supports targeted mitigation strategies based on soil microbiome characteristics.
|
|
Scooped by
mhryu@live.com
Today, 3:54 PM
|
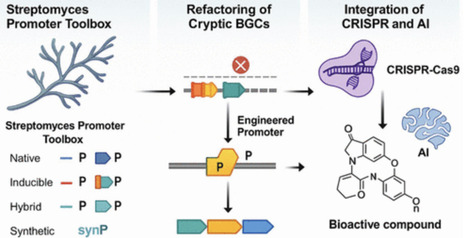
The activation of cryptic biosynthetic gene clusters (BGCs) in Streptomyces has emerged as a powerful strategy to uncover novel antibiotics. Recent advances in native and synthetic promoter engineering have enabled precise transcriptional control over silent gene clusters. This review presents a comprehensive overview of constitutive and inducible promoters, both native and synthetic, used in refactoring efforts. It also highlights CRISPR-based activation systems and machine-learning-guided promoter design for BGC activation. Case studies illustrate how these strategies are translated into enhanced metabolite production, particularly in the context of antimicrobial resistance. This review aims to provide a systems-level understanding of promoter tools and their translational implications for natural product discovery.
|
|
Scooped by
mhryu@live.com
Today, 3:46 PM
|
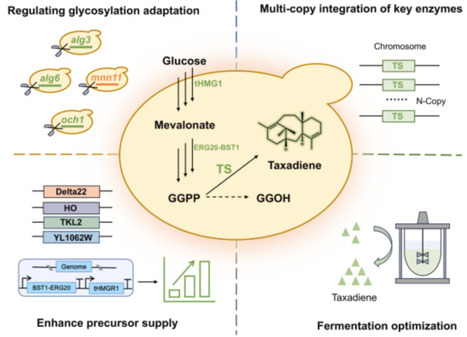
Utilizing synthetic biology techniques to construct recombinant engineered cells for the synthesis of paclitaxel and its key precursors has emerged as an effective method to address the supply–demand imbalance and protect rare plant resources. Taxadiene, a critical precursor of paclitaxel, exhibits limited yield in eukaryotic systems, constraining its biosynthetic potential. Previous research has demonstrated that glycosylation modifications in Saccharomyces cerevisiae substantially impact the regulation of exogenous protein expression. In this study, we found that knocking out endogenous protein glycosylation genes in the chassis significantly improved the expression of heterologous proteins, especially the key taxadiene synthase (TS), and thereby increased the yield of taxadiene. In particular, we identified that the deletion of the glycosyltransferase gene mnn11 increased taxadiene production levels by 65.2 %. The subsequent multi-copy integration of the key enzyme taxadiene synthase further elevated taxadiene production levels in shake flasks by more than 3-fold, reaching 113.5 mg/L. Moreover, the enhancement of geranylgeranyl diphosphate synthesis-related expression modules resulted in a 2.69-fold increase in taxadiene yield, to 420.4 mg/L. Following the optimization of fed-batch fermentation conditions, taxadiene production levels of up to 1.26 g/L were achieved, representing a 63-fold improvement over that obtained with the initial strain. Our findings provide valuable insights into enhancing heterologous taxadiene production efficiency by blocking endogenous protein glycosylation modifications in S. cerevisiae, establishing a critical precedent for improving compatibility between natural product synthesis and microbial cell factories.
|
|
Scooped by
mhryu@live.com
Today, 3:36 PM
|
Glucose oxidase (GOD) is a widely used enzyme in biotechnology, yet its narrow substrate specificity limits its application in complex bioconversion processes such as agricultural waste valorization. In this study, we employed synthetic biology and protein engineering strategies to engineer a broad-spectrum glucose oxidase from Aureobasidium sp. (AreGOD). Initially, site-directed mutagenesis at N82, a key gatekeeper at the dimer interface, modulated substrate channel geometry, leading to increased catalytic activity towards various sugars, particularly stachyose and xylose. Furthermore, systematic linker engineering between the spore anchor protein CotG and AreGOD revealed that flexible linkers, particularly the (GGGGS)5 repeat (LK3), dramatically expanded the enzyme's substrate spectrum towards various mono-, di-, and oligosaccharides. The optimized spore-displayed AreGOD (CotG-LK3-AreGOD) exhibited strong synergistic effects with cellulase in wheat straw degradation, significantly enhancing the hydrolysis of cellulose, hemicellulose, and lignin. Our work demonstrates an effective and generalizable strategy for engineering substrate-promiscuous oxidases, highlighting the potential of integrative enzyme design for sustainable bioprocessing and agricultural biotechnology.
|
|
Scooped by
mhryu@live.com
Today, 3:26 PM
|
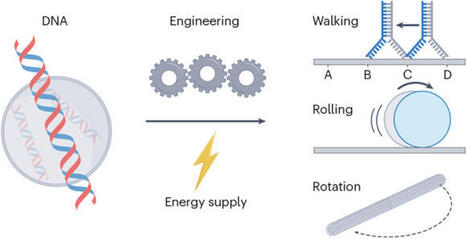
DNA nanotechnology has rapidly evolved, leading to the development of dynamic nanoscale and microscale devices that mimic natural molecular machinery. This Review explores the latest advancements in DNA-based machines, motors and switches, emphasizing the need for clear definitions to distinguish between these often-interchanged terms. By analysing key performance metrics such as speed, force generation, efficiency and autonomy, we provide a framework for evaluating these devices against their biological counterparts, including motor proteins such as myosin and kinesin. We highlight innovative design strategies such as strand displacement, DNA origami and hybrid systems, which enhance the functionality of DNA-based constructs and bridge the gap between synthetic and natural systems. These advancements have promising applications in areas such as targeted drug delivery, biosensing and nanofabrication, although challenges in achieving the high performance and efficiency seen in biological systems remain. Through a synthesis of current research, this Review outlines the opportunities and challenges in the development of DNA-based nanoscale and microscale devices. The development of DNA-based machines is transforming fields such as drug delivery and biosensing. Here, design strategies are discussed and key performance metrics — speed, force generation, efficiency and autonomy — are examined to provide insights into the future of DNA nanotechnology.
|
|
Scooped by
mhryu@live.com
Today, 12:12 PM
|
Natural proteins often form intricate multidomain, oligomeric architectures. This presents a prima facie challenge to cellular homeostasis, as topologically complex proteins seldom refold efficiently in vitro. Here, we show that the efficient folding and assembly of the five-domain homotetramer β-galactosidase is obligatorily coupled to its synthesis on the ribosome, and we define the underlying mechanisms. During refolding from a denaturant, maturation of the catalytic domain is frustrated. Assembly outpaces monomer folding, and non-native oligomers accumulate. Efficient de novo folding is characterized by segmental domain folding, shaped by the binding of a nascent amphipathic helix to a cryptic pocket on uL23 on the ribosome surface. Homomer assembly also initiates cotranslationally via recruitment of a full-length subunit to the nascent polypeptide, and the failure to do so results in misassembly. Our results reveal how the ribosome can dictate the timing of folding and assembly to enable efficient biogenesis of a topologically complex protein.
|
|
Scooped by
mhryu@live.com
Today, 11:58 AM
|
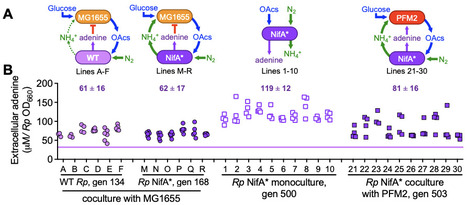
Some microbes externalize costly biosynthetic precursors in sufficient quantities to sustain a recipient population through cross-feeding. However, it is unclear whether metabolites are externalized purely for a reciprocal benefit or if metabolite externalization also plays a physiological role for the producer. Here we focus on adenine, a metabolite externalized by some strains of the phototrophic bacterium Rhodopseudomonas palustris at sufficient levels to support E. coli growth. In 10 long-term monocultures and 22 cocultures pairing R. palustris with E. coli, extracellular adenine externalized by all 140 isolates screened was 1.7 - 3.4-fold higher than that by the ancestor, suggesting that there was selective pressure for adenine externalization. We hypothesized that adenine is toxic to R. palustris. The CGA0092 growth rate decreased by half in the presence of about 0.3 mM external adenine. This inhibitory effect increased by an order of magnitude when we overexpressed adenine phosphoribosyltransferase to overcome a bottleneck in the purine salvage pathway, suggesting that toxicity stems from a metabolite derived from adenine. To assess whether adenine tolerance is connected to adenine externalization, we surveyed 12 evolved isolates and 49 environmental strains that externalized different levels of adenine, revealing a significant positive correlation. Our data suggests a physiological role for externalization of costly-metabolites like adenine at the origin of cross-feeding. In addition to cross-feeding, resulting metabolic interactions could be negative, considering that even a biosynthetic precursor like adenine can be inhibitory.
|
|
Scooped by
mhryu@live.com
Today, 11:45 AM
|
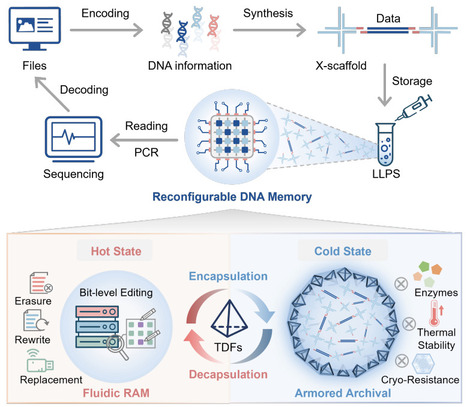
DNA has emerged as a promising medium for the post-silicon era of information storage due to its ultrahigh density and longevity. However, current systems are bifurcated, with solid-state systems providing robust cold archival but lacking accessibility, while fluidic molecular computing systems offer dynamic processing but suffer from low density and instability. This mutual exclusivity has hindered the development of hierarchical memory, a standard in modern computing, within molecular storage systems. Here, we bridge this gap by engineering a reconfigurable DNA memory architecture driven by programmable liquid-liquid phase separation (LLPS). Our system leverages sequence-based encoding to achieve an ultrahigh storage density of 7×10^10 GB/g, approaching the theoretical limits of DNA accessibility. In its fluidic hot state, DNA droplets enable rapid data loading (~83.8% in 5 min) and function as an in-memory editing platform supporting versatile, addressable bit-level operations including selective erasure (~65.1%) and high-efficiency rewriting and replacement (>99%) via programmable strand displacement. Importantly, to resolve the stability trade-off, we engineered a programmable phase transition whereby the triggered assembly of a rigid tetrahedral DNA framework (TDF) armor transforms liquid condensates into robust armored droplets. This cold state confers exceptional resistance to enzymatic and physical degradation, projecting multi-millennial data stability. By enabling reversible transitions between an editable, high-density computing mode and a stabilized archival mode, this work establishes the architectural foundation for scalable molecular information storage capable of hierarchical data management.
|
|
Scooped by
mhryu@live.com
Today, 10:25 AM
|
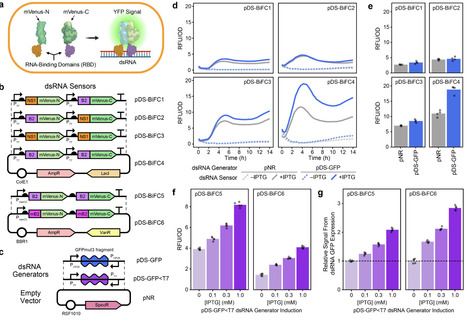
There is growing interest in engineering animal and plant microbiomes to deliver double-stranded RNA (dsRNA) for RNA interference (RNAi) applications. We developed a genetically encoded biosensor that uses bimolecular fluorescence complementation to monitor dsRNA levels within bacterial cells to accelerate the symbiont-mediated RNAi design-build-test cycle. We validated performance of the sensor in Escherichia coli and demonstrated enhanced dsRNA accumulation in engineered strains of the aphid symbiont Serratia symbiotica.
|
|
Scooped by
mhryu@live.com
Today, 10:15 AM
|
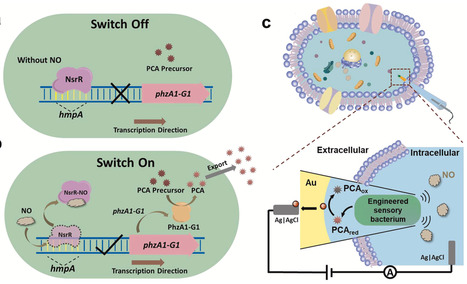
The intracellular gene circuit to transform biochemical signals to an electrical signal is the key for biosensor development to gap the information mismatch at the bio-electric interface, but a qualified gene circuit is difficult to design. In this study, we have shown the construction of an integrated synthetic gene circuit including the nitric oxide (NO)-responsive module and the phenylenediamine-1-carboxylic acid (PCA) synthesis module, to (1) equip a less renounced electroactive bacterium (e.g. Escherichia coli) with indirect electron transfer (IET) pathway to enhance electrical signal output, as well as (2) couple the IET gene circuit with the responsive gene circuit to sense small signal molecule, NO. The subsequent oxidation of PCA can be electrochemically quantified by the microelectrode, thereby establishing a signaling pathway from intracellular message to redox mediator, and finally to electrical signal output. In this way, the constructed microbial electrochemical biosensor for intracellular in situ NO analysis at the single-cell level owns high sensitivity, a wide linear detection range (100–2500 nM), and excellent selectivity. Therefore, this work shows an example for developing next-generation electroactive microorganisms (EAMs)-based electrochemical biosensors through synthetic biology tools with tailored and intelligent functionalities.
|
|
Scooped by
mhryu@live.com
January 18, 12:42 PM
|
Teosinte (Zea mays subsp. mexicana) has been proposed as a potential source of biological nitrification inhibition (BNI), yet how nitrogen (N) inputs modulate its exudate chemistry and associated nitrification processes remains unclear. We compared teosinte with three maize cultivars under N-deficient and N-replete conditions, integrating non-targeted metabolomics of root exudates, qPCR of rhizosphere amoA genes, and pure-culture assays with Nitrosomonas europaea. N fertilization enhanced total root exudation and reprogrammed the teosinte metabolome toward amino and phenolic acids, with histidine, glutamic acid, ferulic acid, and vanillic acid being markedly enriched. These compositional shifts coincided with reduced archaeal amoA abundance in teosinte (and Zhengdan958) but increased levels in Ye478 and Qi319. In culture, exudates from N-fed teosinte strongly inhibited N. europaea ammonia oxidation (~ 63%), whereas exudates from modern maize, except for Zhengdan958, showed little effect. Histidine, vanillic acid and ferulic acid reproduced inhibition in targeted assays, implicating them as candidate BNIs likely acting through copper chelation and phenolic interference. Collectively, these findings demonstrate that N availability reshapes teosinte exudate chemistry, thereby strengthening nitrification suppression through specific amino- and phenolic-acid release. Leveraging these wild traits could inform sustainable N management and enhance nitrogen-use efficiency in maize-based agroecosystems.
|
|
Scooped by
mhryu@live.com
January 18, 12:18 PM
|
Oral live microbial therapeutics (LMTs) show promise for halitosis, caries, and adjunctive periodontal care, yet benefits often fade after dosing stops. We synthesized evidence across indications and reframed development around quantifying and engineering persistence at intraoral sites, while outlining safety-by-design and delivery considerations for the oral niche. This narrative review integrated randomized trials, observational studies, and in vitro/ex vivo investigations to characterize clinical outcomes, persistence-related metrics, and engineering principles relevant to oral LMT development. Sources included PubMed/MEDLINE, Web of Science, Embase, and ClinicalTrials.gov, with backward/forward citation tracking. We included studies on LMTs in oral or gut contexts when mechanistically informative for oral applications (e.g., persistence, delivery, or biocontainment). Eligibility required clinical outcomes or persistence-related readouts. Two reviewers screened records and resolved disagreements by consensus. Reporting and assay principles were informed by STORMS and MIQE to support transparent, reproducible methods. Across indications, effects typically peak during dosing and attenuate after cessation, varying with strain, delivery format, and co-interventions (e.g., tongue dorsum debridement; standardized periodontal care). Persistence is rarely co-measured with clinical endpoints, limiting mechanistic interpretation. We outline a site-resolved measurement set, including time above the limit-of-detection, colonization area under the curve, apparent half-life (t½), and t½ under oral-mimetic shear, together with an engineering toolkit combining mucoadhesive/enamel-interactive carriers, single-cell coatings, and multilayer biocontainment (e.g., logic-gated/CRISPR kill switches, synthetic auxotrophy), and chemistry, manufacturing, and controls considerations. Embedding persistence metrics and safety-by-design into study protocols may support more durable outcomes, and standardized, site-resolved reporting will be essential for clinical translation.
|


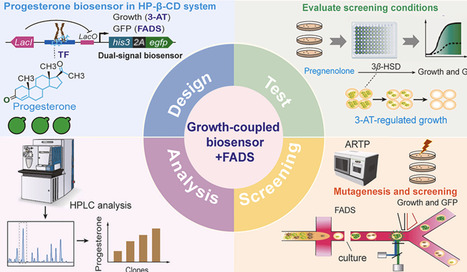



















expressing EGFP for fluorescence readout and HIS3 for growth-based selection via a self-cleaving 2A peptide. Progesterone produced from pregnenolone by 3β-HSD activates a synthetic LexA–PRO–VP16 transcription factor, coupling metabolite formation to measurable outputs. IPTG induction and 3-AT supplementation allow tuning of selection stringency.
To minimize background growth and reduce false positives, 3-AT, a competitive HIS3 inhibitor, was employed. S077 exhibited basal growth even in the absence of progesterone, consistent with low-level HIS3 expression. The addition of 2.5 mM 3-AT effectively suppressed this background growth, enhancing sensor specificity.