 Your new post is loading...

|
Scooped by
mhryu@live.com
Today, 4:33 PM
|
Over the past few decades, antibacterial discovery has changed substantially, yet innovation has remained slow despite the growing need for new agents active against multidrug-resistant pathogens. Progress is constrained by scientific hurdles, including target selection, penetration and efflux in bacteria, and the difficulty of translating in vitro activity into in vivo efficacy, as well as persistent economic disincentives. Here, we discuss these challenges and summarize recent advances in the field, with a medicinal chemistry focus on synthetic small molecules that have reached validated-lead or preclinical development and shown in vivo antibacterial efficacy. We also provide an outline of the current clinical pipeline, highlighting areas of innovation and remaining gaps. Our goal is to offer a comprehensive perspective on antibacterial discovery that supports ongoing efforts to strengthen the pipeline in response to antimicrobial resistance.
|
|
Scooped by
mhryu@live.com
Today, 4:12 PM
|
The architecture of Bacillus subtilis biofilms is influenced by the coordinated regulation of cellular specialization, matrix assembly, and metabolism. B. subtilis can form different types of biofilm in diverse physical and chemical environments. Understanding the molecular mechanisms that drive biofilm heterogeneity and adaptation to different environmental niches is crucial for developing more effective strategies to control their formation. In this study, we developed a tightly dual-regulated CRISPR interference (CRISPRi) system and employed multi-scale imaging to investigate the functions of individual genes in two distinct biofilm models: the floating pellicle and the intricate, three-dimensionally structured macrocolony, which develop at the liquid-air and solid-air interfaces, respectively. Our findings validated the CRISPRi approach as a powerful method for studying biofilm development over extended periods and revealed that numerous small non-coding RNAs are involved in regulating biofilm growth dynamics and architecture. The CRISPRi approach was also applied to a pool of 507 genes and transcription units, including protein-coding genes and non-coding RNAs, to screen for cell fitness in these two biofilm models. We discovered that, while both biofilm forms rely on fundamental processes such as cell wall synthesis and nucleotide metabolism, they exhibit different genetic dependencies with regard to matrix composition, motility, and signaling. Exopolysaccharide production, motility, and chemotaxis are crucial for pellicle formation. In contrast, macrocolony development is influenced by γ-polyglutamate synthesis and nutrient acquisition functions. Genes of unknown function were also identified to play a differentially important role in the two biofilm forms. Additionally, the CRISPRi screens revealed further non-coding RNAs regulating biofilm architecture and growth dynamics, adding to the existing layers of post-transcriptional control. Collectively, these results demonstrate that biofilm formation at different physical interfaces is governed by a combination of shared and unique genetic pathways tailored to the specific biofilm environment, thereby opening research avenues into the molecular mechanisms specific to the solid-air and liquid-air interfaces.
|
|
Scooped by
mhryu@live.com
Today, 3:54 PM
|
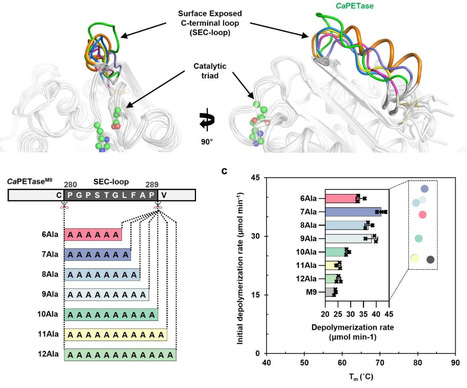
Polyethylene terephthalate (PET) hydrolases have been extensively studied for their potential applications in plastic degradation. However, the structural and mechanistic factors that limit their catalytic efficiency are not yet fully understood. Here, we identify the protruding, surface-exposed C-terminal loop (SEC-loop) in Cryptosporangium aurantiacum PETase (CaPETase) that negatively impacts enzymatic activity by restricting productive access of enzyme to PET. Loop replacement experiments show the non-protruding SEC-loop enhances PET depolymerization rates, despite being ~25 Å from the active site. Kinetic and adsorption studies indicate the non-protruding SEC-loop promotes productive PET access to the enzyme without affecting binding affinity. To further assess the broader applicability of this strategy across diverse PETases, SEC-loop replaced variants of representative PETases are characterized through kinetic and adsorption analyses. We show an engineering strategy focused on modulating enzyme accessibility rather than simply modifying the catalytic site, in rational enzyme design aimed at improving PET degradation efficiency. Polyethylene terephthalate (PET) hydrolases have been extensively studied for their applications in plastic degradation, but the structural and mechanistic factors that limit their catalytic efficiency are not yet fully understood. Here, the authors identify the protruding, surface-exposed C-terminal loop (SEC-loop) in Cryptosporangium aurantiacum PETase that negatively impacts enzymatic activity by restricting productive access of enzyme to PET substrates.
|
|
Scooped by
mhryu@live.com
Today, 3:16 PM
|
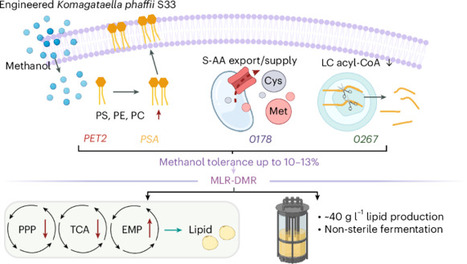
Methanol is a promising C1 feedstock for bioproduction but its cytotoxicity limits applications. Here we enhanced Komagataella phaffii GS115 methanol tolerance to 10–13% (v/v) by overexpressing four genes (phosphatidylethanolamine methyltransferase (PET2), phosphatidylserine synthase (PSA), PAS_chr1-4_0178 (0178) and PAS_chr2-2_0267 (0267)) (S33 strain). Our results show that, under high methanol stress, S33 undergoes membrane lipid remodelling-driven metabolic reprogramming, with central carbon flux being redistributed from the pentose phosphate pathway and citric acid cycle towards glycolysis. Within this framework, 0178 encodes a vacuolar transporter of sulfur-containing amino acids, which we propose supports phospholipid methylation and redox balance. Meanwhile, 0267 encodes a peroxisomal thiolase-degrading long‑chain fatty acyl‑CoAs, limiting toxic long-chain sphingolipid accumulation, maintaining membrane stability. These genes enable S33 to achieve lipid titre of approximately 40 g l−1 under non-sterile fed‑batch fermentation conditions. This work positions S33 as a methanol-tolerant K. phaffii chassis for further strain and process development. Bioproduction from methanol is often challenging owing to the cytotoxicity of methanol. Here a methanol-tolerant Komagataella phaffii chassis is engineered by overexpressing four membrane genes, increasing tolerance to 10–13% methanol and enabling non-sterile lipid production (>40 g l−1) via membrane lipid remodelling and metabolic rewiring.
|
|
Scooped by
mhryu@live.com
Today, 3:07 PM
|
Designing multichain protein complexes requires coordinating the folding of component proteins with the formation of their interfaces. The existing methods, however, remain limited in their ability to satisfy these requirements simultaneously, especially for trimeric and tetrameric complexes. As an important practical scenario, designing a binder that bridges two target proteins into a ternary complex requires flexibility in the relative arrangement of the two targets, adding an additional challenge to existing design methods. Results: We present ComplexDesign, a hallucination-based approach for multichain protein design. ComplexDesign performs structure-prediction-guided sequence optimization to simultaneously fold each protein chain and form inter-chain interactions that bind them together. To provide the flexibility required to appropriately arrange these target proteins, ComplexDesign introduces a specialized masking mechanism that enables exploration of possible relative arrangements rather than being limited to the predefined ones. Across a comprehensive set of benchmarks with various chain lengths, ComplexDesign outperformed existing methods in the unconditional design of dimers, trimers, and tetramers, achieving a high design success rate exceeding 50%, supporting its capability for multichain complex design. Furthermore, in the case of multi-target binder design, ComplexDesign produced high-confidence, self-consistent ternary complexes for 8 out of 10 target pairs. These results establish ComplexDesign as an effective tool for multichain protein design, with particular utility for designing binders that bridge two target proteins. Availability and implementation: The source code of ComplexDesign will be made publicly available upon publication.
|
|
Scooped by
mhryu@live.com
Today, 11:24 AM
|
The design of proteins that bind to small molecules has been challenging because it requires simultaneous optimization of the protein sequence, protein structure and ligand conformation. Current deep-learning algorithms have struggled to navigate this landscape, precluding the zero-shot design of binders. Here we show that by combining two neural networks in an iterative design algorithm, small-molecule binding proteins can be created from scratch with high accuracy. We trained a graph neural network—ligand-aware sequence engineering message-passing neural network (LASErMPNN)—to design compatible protein sequences for an input protein backbone and docked ligand. We paired LASErMPNN with a structure predictor that models a three-dimensional protein–ligand complex for an input protein sequence and ligand identity. The closed-loop iteration of these reciprocal networks optimized sequence–structure–ligand compatibility, and outperformed a comparable design loop using a physics-based energy function. We used our strategy, termed neural iterative selection–expansion (NISE), to design proteins that, using different folds, specifically bind to two chemically distinct small-molecule drugs, exatecan and apixaban, with success rates of 100% and 83%, respectively. The tightest NISE binders had nanomolar-to-picomolar affinities, surpassing those of the next-leading method by 70-fold for exatecan and nearly 10,000-fold for apixaban. LASErMPNN then suggested two amino-acid substitutions that improved the affinity of the tightest exatecan binder by 100-fold without any experimental input. The optimized binder protected the labile lactone ring of exatecan from hydrolysis for days. Our work describes a general recipe for using neural networks to automate the design of small-molecule binding proteins for applications in drug delivery, sensing and catalysis. By pairing two neural networks in an iterative optimization algorithm, small-molecule binding proteins can be designed from scratch with high accuracy, affinity and success rates, showing promise for applications in drug delivery and sequestration.
|
|
Scooped by
mhryu@live.com
Today, 12:44 AM
|
The term ‘gut health’ is increasingly used as a catch-all phrase by many stakeholders, including scientists, health-care professionals, industry and the general public, to describe a wide range of health-related concepts. Despite its widespread use, particularly in relation to studies on diet, fermented foods, biotics and the gut microbiome, it remains unclear what the term gut health means. Therefore, an expert panel was convened by the International Scientific Association for Probiotics and Prebiotics to address the current state of scientific and clinical knowledge on the physiology, manifestation, application and measurement of the concept of gut health. The panel evaluated the term in the context of the central role of the gastrointestinal tract in health and overall well-being and proposed a definition of gut health as “a state of normal gastrointestinal function without active gastrointestinal disease and gut-related symptoms that affect quality of life”. The definition was developed mindful of the functional, subjective and extrinsic domains that contribute to gut health. In this Consensus Statement, clinically relevant and accessible metrics to assess these domains are reviewed and a comprehensive approach to gut health is proposed that is relevant to clinical practice as well as to studies of dietary and biotic interventions. This Consensus Statement provides a definition of the term ‘gut health’, as well as a discussion of the relevant domains that contribute to gut health and a framework for appropriate use of the term in the context of therapeutic interventions.
|
|
Scooped by
mhryu@live.com
Today, 12:35 AM
|
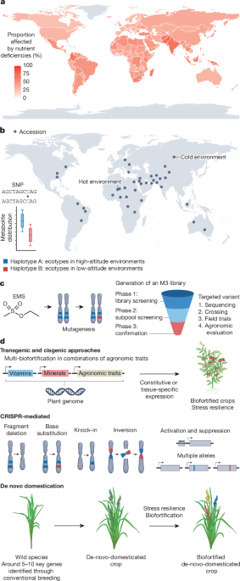
At present, more than 700 million people live with caloric hunger, and more than two billion suffer from micronutrient deficiencies, known as ‘hidden hunger’. From an agricultural viewpoint, three major objectives need to be worked towards simultaneously to achieve zero hunger (the United Nations Sustainable Development Goal 2): (1) enhanced yield; (2) higher vitamin and mineral density to sustain recommended daily intake (multi-biofortification); and (3) enhanced climate-change resilience. Although the Green Revolution increased global calorie production, it exacerbated hidden hunger by prioritizing high yield over nutritional quality. Stress from global climate change has been shown to reduce the densities of several micronutrients. CRISPR–Cas, which allows genome editing with extremely high precision, has emerged as a groundbreaking breeding technology that has already been adopted by many countries. Here we examine how CRISPR–Cas-based approaches could be used to achieve biofortification targets by enhancing micronutrient densities to the levels necessary to alleviate dietary vitamin and mineral deficiencies. Given the limited time frame available to achieve zero hunger, we argue that CRISPR–Cas technologies should be combined with metabolic engineering based on transformation and other technologies. We also consider untapped resources beyond metabolic pathways and current CRISPR–Cas methodologies to address one of the most important societal issues of the twenty-first century. This Review reflects on the joint power of genetic technologies, including untapped CRISPR–Cas techniques to combat hidden hunger and improve crop resilience, and argues in favor of their combined use to overcome these societal challenges.
|
|
Scooped by
mhryu@live.com
June 24, 12:36 PM
|
Structure-based virtual screening (VS) via molecular docking is a pivotal approach for hit identification. Many artificial intelligence (AI)-powered protein–ligand docking and scoring methods have demonstrated impressive speed and accuracy. Retrospective benchmarking studies using enrichment rate and computational efficiency on curated datasets have corroborated their potential for discovering bioactive compounds. However, determining which method suits a specific application and implementing it efficiently remains challenging. Here we present the Comprehensive VS Platform with AI Engine (CVSP-AIE) for drug discovery from compound libraries. It integrates three AI models: KarmaDock, a fast docking model that directly updates atomic coordinates; CarsiDock, an accurate docking model that predicts protein–ligand distances and reconstructs binding poses; and RTMScore, an accurate scoring model that learns residue–atom distance distributions for affinity prediction. Their hierarchical application enables dynamical balances in screening speed and accuracy. CVSP-AIE is available as an online web server ( https://cadd.zju.edu.cn/cvsp/ ) and a local software package. Users can efficiently initiate drug screening by uploading a protein and a known binder that defines the binding pocket. The following workflow involves (1) preprocessing, including protein structure repair and molecule standardization, (2) binding pose and affinity prediction powered by KarmaDock, CarsiDock and RTMScore and (3) postprocessing, comprising protein–ligand interaction calculation and visualization. It takes 30–45 min to hierarchically screen 100,000 compounds, and the output is a ranked list of molecules with predicted binding scores, intermolecular interaction profiles and interactive chemical space analysis. Users can also install locally the hierarchical screening module through command-line package for arbitrary-scale screening. This Protocol describes an artificial intelligence-driven virtual screening platform for drug discovery that includes an online webserver and a local software package, offering a user-friendly alternative for drug screening targeting compound libraries of arbitrary-scale.
|
|
Scooped by
mhryu@live.com
June 24, 12:07 PM
|
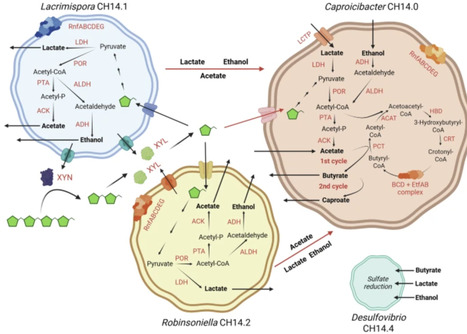
Lignocellulose is a promising renewable resource for anaerobic biochemical production, but its microbial conversion remains challenging. To elucidate metabolic networks in lignocellulose-degrading consortia, inocula of various origins were enriched on cellulose or xylan. Community composition and metabolic functions were revealed by amplicon sequencing, metagenomics, genome-scale metabolic modelling, and metabolic simulations. In cellulose-enriched communities, Fibrobacter and Lacrimispora consistently dominated as primary cellulose degraders, whereas Bacteroides likely functioned as secondary degraders. Acetic acid (up to 1.3 g l-1) and CO2 were the main fermentation products. Xylan enrichments produced C2-C6 fatty acids (up to 3.9 g l-1), lactic acid (up to 1.2 g l-1), ethanol (up to 1.2 g l-1), CO2, and H2. Clostridium dominated one xylan community and produced mainly butyric acid, while Bifidobacterium dominated another and produced mainly lactic acid. Caproic acid production was experimentally observed in one xylan enrichment. Metagenomic annotations and metabolic simulations suggest that Lacrimispora amygdalina degraded xylan and Robinsoniella peoriensis consumed xylobiose as a secondary consumer, both likely producing ethanol and lactic acid that supported caproic and butyric acid production by Caproicibacter fermentans. Integrated analysis identified functional guilds and clarified the roles of degraders and non-degraders, providing a blueprint for engineering synthetic consortia for sustainable biochemical production.
|
|
Scooped by
mhryu@live.com
June 24, 11:58 AM
|
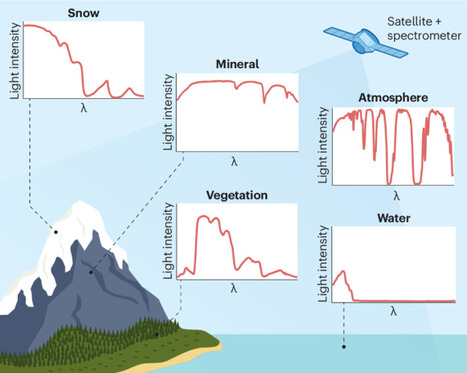
Imaging spectroscopy technology is transforming the way Earth is viewed from space, with applications across diverse science communities. A global imaging spectrometer mission with Landsat-like spatial and temporal coverage could fully realize this potential.
|
|
Scooped by
mhryu@live.com
June 24, 11:54 AM
|
Pseudomonas putida KT2440, renowned for its diverse metabolic capabilities, is a promising platform for downstream processing and revalorisation of recalcitrant molecules. In this study, we examined and optimized P. putida KT2440's ability to utilize products of the degradation of polyethylene (PE), the most used and disposed plastic. PE degradation creates over 200 molecules that vary in oxidation level and, thus, chemical properties. Among those, long-chain alcohols represent one of the most challenging fractions to process due to their poor solubility. Using them as feedstock for microbial growth would close the plastic-derived carbon cycle, reducing environmental impact. First, we discovered that P. putida KT2440 can use the long-chain alcohols, 1-hexadecanol and 1-eicosanol, as the sole carbon and energy source. Using adaptive laboratory evolution (ALE), we generated variants with improved growth rates on such substrates. Mutations that became fixed during ALE provided insights into the mechanism, highlighting the importance of cell–substrate interaction. By heterologously expressing a hydrocarbon transporter-encoding gene, we successfully reproduced the ALE-derived phenotype, suggesting that the bottleneck in long-chain alcohol utilisation lies in uptake rather than substrate transformation. These findings lay the groundwork for the potential application of P. putida KT2440 for the valorization of PE degradation products.
|
|
Scooped by
mhryu@live.com
June 24, 11:48 AM
|
Cereal root microbiomes harbor diverse diazotrophic bacteria, yet the taxa capable of sustained nitrogen fixation in association with cereal roots remain poorly characterised. Here, two high-performing nitrogen-fixing strains (B6 and J2) were isolated from barley roots and identified as belonging to the family Rhizobiacae in the genus Paenirhizobium. Both strains possess plasmid-encoded canonical rhizobial nif and fix genes for nitrogen fixation but lack nodulation genes. Their genomes have a 5.7 Mb chromosome and four repABC plasmids. Unlike most nodulating rhizobia, strains B6 and J2 fixed nitrogen in laboratory culture on a range of carbon sources, achieving maximal activity on organic acids at low ammonium (<0.5 mM) and oxygen concentrations (1–3%). Both strains colonized the total root systems of barley plants, with population densities of 106 CFU g−1 fresh root weight. Strains fixed high levels of nitrogen on barley plants, similar to or greater than other known free-living diazotrophs. These findings expand the ecological context of rhizobial nitrogen fixation and identify cereal-associated Paenirhizobium as a previously unrecognised component of the diazotrophic cereal root microbiome.
|
|
|
Scooped by
mhryu@live.com
Today, 4:20 PM
|
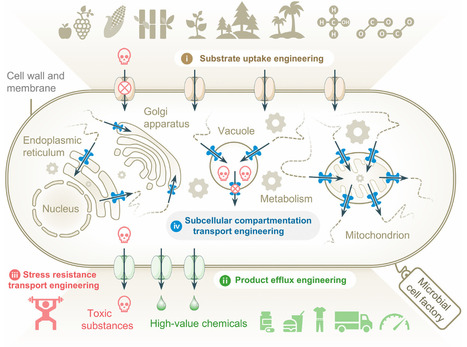
Microbial cell factories are central platforms for sustainable industrial biomanufacturing, yet inefficient transmembrane transport continues to hinder their large-scale industrial translation. Transporter engineering has progressed from trial-and-error overexpression to mechanism-guided rational design, offering effective routes to relieve transport-related metabolic constraints. This review summarizes key advances in substrate uptake, product efflux, stress tolerance, and subcellular compartmentalized transport, analyzes evolutionary trade-offs and practical engineering bottlenecks of transporters, and reviews applications of omics, high-throughput screening, and AI. It aims to deliver a systematic overview of the field and support the further development of transporter engineering for robust industrial microbial cell factories that enable efficient and stable production of high-value chemicals in next-generation green and sustainable biological manufacturing systems.
|
|
Scooped by
mhryu@live.com
Today, 4:05 PM
|
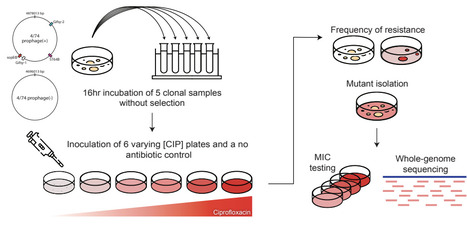
Most naturally occurring bacteria are lysogens, encoding one or more temperate phages integrated into their genome. As prophages are induced by the bacterial SOS response, DNA-damaging antibiotics can trigger SOS-mediated prophage induction, where prophages undergo lytic replication and lyse their host, even at sub-inhibitory concentrations. This prophage-antibiotic synergy therefore sensitizes lysogenic hosts to DNA-damaging antibiotics. However, the mechanism by which prophage-induced sensitization affects the evolution of resistance against these agents is unclear. Here we show that ciprofloxacin-resistant lysogens arise less frequently but exhibit higher levels of resistance following selection. Whole-genome sequencing showed that increased lysogen resistance arose from selection towards mutations in drug targets, efflux pathways, and stress response regulators that reduce antibiotic efficacy or alter SOS induction. Consistent with this result, resistant lysogens exhibited a dampened SOS response, suggesting that prophage induction imposes an additional selective filter on their hosts by eliminating mutants that experience sufficient DNA damage to activate the SOS response. By contrast, prophage carriage had no effect on sensitivity or resistance evolution for antibiotics where DNA damage occurs downstream of the primary mechanism of action. Together, these findings indicate that prophage induction acts as an evolutionary bottleneck that restricts many resistance trajectories while favoring the emergence of rarer, large-effect mutations, potentially accelerating the evolution of high-level resistance.
|
|
Scooped by
mhryu@live.com
Today, 3:21 PM
|
Bitter taste is a critical quality determinant in food systems, particularly those using sustainable protein hydrolysates, where the unpredictable formation of bitter peptides severely limits consumer acceptance. Achieving predictive control over flavor chemistry requires deciphering the complex sequence-activity relationship. To address this, we integrated the generative capacity of a protein language model with BitterPep-GCN, a Graph Convolutional Network (GCN) capable of robust in silico bitter/non-bitter classification, to target the de novo design of functional bitter and non-bitter sequences. We achieved this by generating two strategic peptide libraries: a targeted tripeptide library derived from known bitter and non-bitter peptide sequences, and a set of de novo designed sequences. For the de novo designed peptides, we fine-tuned the conditional language model ZymCTRL on our curated dataset of sensory-validated bitter peptides (BPS-1000). Both libraries were subjected to classification and rigorous filtering using BitterPep-GCN to select high-confidence candidates for validation. The selected peptides were purchased and rigorously assessed for high purity. Sensory tests were conducted by an expert human panel to determine intrinsic taste quality and taste recognition thresholds. The results validated the high predictive fidelity of our pipeline: out of the 31 tested peptides, 25 were correctly classified, including 15 confirmed bitter and 10 confirmed non-bitter sequences. This study successfully demonstrates the application of machine learning frameworks in the design of bioactive peptides. It provides a set of novel taste-active peptides that can be used to accelerate the rational mitigation of off-tastes in next-generation food products.
|
|
Scooped by
mhryu@live.com
Today, 3:14 PM
|
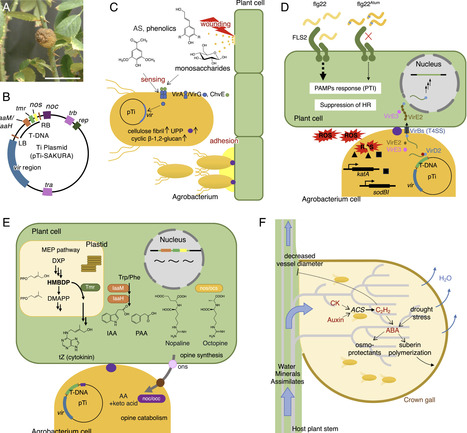
Plants and pathogens engage in complex biochemical communication, mediated by proteins, specialized metabolites, phytohormones and phytohormone-mimicking compounds. These interactions drive a dynamic ‘co-evolutionary arms race’, as plants and microbes compete to gain an advantage, ultimately transforming the infection site into a distinct micro-ecosystem. Microbe-induced galls are abnormal plant organogenesis induced by specific pathogens, creating a niche where the pathogen manipulates the host plant machinery to ensure its own survival. Such galls adversely affect agricultural and horticultural productivity by stunting plant growth and causing deformities. However, the rapid cell proliferation in tumourigenesis, along with the mechanisms driving robust shoot and root re-differentiation, present exciting opportunities for innovative biotechnological applications. Therefore, elucidation of these mechanisms is crucial for advancing basic research with significant potential for agricultural applications. This review focuses on galls induced by Agrobacterium tumefaciens and Rhodococcus fascians—two phytopathogens that utilize phytohormones as tumor-inducing molecules—to highlight the mechanisms underlying plant–pathogen interactions within this specialized microenvironment. It also explores the evolutionary adaptations and strategies of these pathogens. Gaining insight into these biological processes is key to understanding the mechanisms driving biological diversity and evolution, with implications extending beyond plant pathology into the broader field of molecular plant physiology.
|
|
Scooped by
mhryu@live.com
Today, 12:03 PM
|
Amino acid substitutions may substantially alter protein stability and function. However, the contribution of substitutions that arise from alternate translation (deviations from the genetic code) is unknown. Here to address this issue, we analysed deep proteomic, transcriptomic and genomic data from more than 1,000 human samples, including 6 cancer types and 26 healthy human tissues. This global analysis identified 60,803 fragmentation spectra corresponding to 8,746 unique substitutions in proteins derived from 1,767 genes, including 1,955 confidently localized sites. Some substitutions were shared across samples, whereas others exhibited strong tissue-type and cancer specificity. Notably, products of alternate translation were more abundant than their canonical counterparts for hundreds of proteins, which suggests that there is sense-codon recoding. Recoded proteins included transcription factors, proteases, signalling proteins and proteins associated with neurodegeneration. Mechanisms that contribute to substitution abundance included protein stability, codon frequency, codon–anticodon mismatches and RNA modifications. We also characterized how alternatively translated proteoform ratios vary across protein domains, tissue types and cancers. These ratios were positively associated with intrinsically disordered regions and genetic polymorphisms in the gnomAD database, although the polymorphisms could not account for the substitutions. The sequence, relative abundance and the tissue specificity of alternatively translated proteins were conserved between humans and mice. These results demonstrate the contribution of alternate translation to the diversification of mammalian proteomes and its association with protein stability, tissue-specific proteomes and disease. Alternate RNA decoding, an understudied process, leads to peptide sequence modifications that can have substantial functional effects on protein stability, tissue-specific proteomes and disease.
|
|
Scooped by
mhryu@live.com
Today, 12:51 AM
|
Polycyclic aromatic hydrocarbons (PAHs), especially high-molecular-weight PAHs (HMW-PAHs), are persistent environmental pollutants that are difficult to remove and challenge environmental management. Altererythrobacter sp. H2 degrades benzo[a]pyrene, benzo[a]anthracene, pyrene, fluoranthene, and phenanthrene, showing great potential in HMW-PAH removal. The aerobic degradation of HMW-PAHs by this strain is initiated by those well-studied ring-hydroxylating oxygenases, followed by subsequent dehydrogenation by less-studied dehydrogenases. Therefore, we investigated the substrate range and substrate recognition mechanism of dehydrogenase PahB from strain H2. Here, we found that PahB from Altererythrobacter sp. H2 can oxidize different HMW-PAH-derived dihydrodiols. Phylogenetic analysis showed that PahB belongs to the NahB-type branch, where it clusters with HMW-PAH-associated homologs, such as BphB-CHY-1, and remains distinct from biphenyl-type BphB dehydrogenases. Crystal structures and docking analyses revealed a hydrophobic, methionine-rich substrate-binding pocket that accommodates different HMW-PAH-derived dihydrodiols in a similar manner. Mutagenesis further showed that methionine residues in this pocket contribute to substrate binding and catalysis. Together, these results define the structural and sequence basis for PahB activity toward HMW-PAH-derived dihydrodiols and expand our understanding of the downstream catabolism of carcinogenic HMW-PAHs.
|
|
Scooped by
mhryu@live.com
Today, 12:41 AM
|
Plants interact with a vast variety of microbes that inhabit both above- and belowground tissues. Through their effect on host physiology and growth, plant-microbe interactions define the success of a plant’s life cycle. A key aspect of these interactions is the requirement for highly cell-type-specific responses from the plant, be it to form symbiotic structures in certain cells or to mount a highly localised immune response. There has been long-standing interest in uncovering the cell-specific transcriptomic changes that underpin these processes to better understand the establishment, functioning, and regulation of plant-microbe interactions. The recent optimization of single-cell and spatial transcriptomics for plants now allows us to investigate these interactions in unprecedented detail. Here, we discuss how single-cell technologies can help unravel the many mysteries of plant-microbe interactions. We focus on the key lessons we have learned from recent single-cell studies in the field and highlight the current limitations of single-cell technologies. We also offer promising avenues for future exploration and conclude by suggesting experimental and bioinformatic considerations to maximize insights from past and future studies and help make the most of this new single-cell era in the field of plant-microbe interactions.
|
|
Scooped by
mhryu@live.com
June 24, 1:01 PM
|
Natural products remain a major source of antibiotics, but discovery efforts have traditionally treated biosynthetic gene clusters as sources of individual bioactive molecules. Increasing evidence has suggested that microorganisms can instead encode coordinated multi-metabolite systems, yet the genetic architectures and biological logic of such systems remain poorly understood. Here we show that Streptomyces spp. encode a highly conserved biosynthetic megacluster that produces four structurally distinct natural product families—stravidins, acidomycin, dapamycins, and 2-methyl-7-keto-8-aminopelargonic acid (α-Me-KAPA)—alongside the biotin-binding protein streptavidin. These components converge on bacterial biotin metabolism through complementary mechanisms, including enzyme inhibition, prodrug activation, cofactor mimicry and biotin sequestration. The encoded metabolites are co-produced and act synergistically across Gram-negative and mycobacterial species, with stravidin S2 and α-Me-KAPA showing enhanced efficacy in combination in a mouse model of multidrug-resistant E. coli infection. This megacluster reveals a genetically encoded chemical arsenal that functions as a naturally evolved combination therapy against a conserved metabolic pathway. More broadly, our findings suggest that higher-order biosynthetic architectures may represent an overlooked reservoir of antibiotic mechanisms and support a shift from discovering isolated natural products to reconstructing native synergistic systems. In Streptomyces spp., a conserved biosynthetic gene megacluster produces an arsenal of distinct antimicrobials that converge on bacterial biotin biosynthesis as a naturally evolved combination therapy.
|
|
Scooped by
mhryu@live.com
June 24, 12:25 PM
|
The cross-species delivery of megabase-scale synthetic DNA molecules, from microorganisms into mammalian cells, remains a major challenge for synthetic genomics. Recently, we developed nucleus isolation for chromosome extraction (NICE), a method that enables the isolation of yeast nuclei containing intact synthetic megabase-scale DNA with preserved chromatin structure. By leveraging the unique epigenomic features of Saccharomyces cerevisiae, which lacks cytosine methylation and repressive histone marks, synthetic DNA encapsulated within isolated yeast nuclei was successfully delivered into mouse early embryos, maintaining a naive state. This work established a unique platform for studying the establishment of de novo epigenetic modifications and their influence on transcriptional regulation over time. Here, we provide a detailed protocol for NICE, including the isolation of yeast nuclei and their subsequent delivery into mammalian embryos. The high-concentration and high-purity isolated nuclei can be stored at –80 °C for >6 months. Using microinjection, we achieved 100% delivery efficiency, reliably transferring isolated yeast nuclei into mouse embryos. The entire procedure, including pulsed-field gel electrophoresis verification, can be completed within ~5 d. When the isolated yeast nuclei are intended for cross-species delivery into embryos, prior familiarity with mammalian embryo microinjection techniques may be required. This protocol offers an efficient and reliable method for the delivery of large-scale genetic information, advancing the study of complex biological functions. This protocol presents a method for isolating intact yeast nuclei containing megabase-scale synthetic DNA and delivering them into mouse embryos, enabling efficient cross-species transfer and studies of de novo epigenetic regulation.
|
|
Scooped by
mhryu@live.com
June 24, 12:01 PM
|
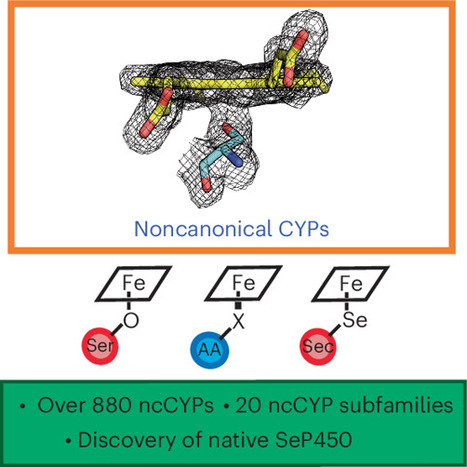
Cytochrome P450s (CYPs) constitute a superfamily of thiolate-ligated heme metalloenzymes principally responsible for the hydroxylation of unactivated C–H bonds. The proximal cysteine is an obligatory and universally conserved residue for the CYP enzyme class. Herein, we challenge this paradigm by systematically identifying noncanonical CYPs (ncCYPs) that do not harbor a proximal cysteine ligand. Our bioinformatic search revealed 20 distinct ncCYP families encoded in diverse microbial genomes with alternative residues at this position. We characterize a native serine-ligated CYP with a high-spin ferric resting state that catalyzes azide reduction and nitrene insertion reactions. Its crystal structure clearly shows a typical CYP fold and a serine alkoxide as a proximal heme ligand. In addition, we report the discovery and characterization of the first native selenocysteine-ligated CYP in nature. Our findings expand the CYP metalloenzyme family and provide opportunities for future enzymatic and biocatalytic discoveries. Cytochrome P450s catalyze essential reactions and carry a strictly conserved proximal cysteine ligand. Here, we identify noncanonical P450s that harbor diverse proximal ligands, including serine and selenocysteine, expanding the P450 chemical space and providing opportunities for future discoveries.
|
|
Scooped by
mhryu@live.com
June 24, 11:58 AM
|
This study addresses the core challenge of Fusarium wilt control in agricultural production. We successfully reconstituted a functional heterologous type III secretion system (T3SS) from Photorhabdus luminescens in the biocontrol bacterium Pseudomonas protegens Pf-5, creating an engineered molecular syringe for targeted delivery of antifungal effectors. The system is activated under low-calcium conditions, achieved by cultivation in calcium-limited medium followed by EGTA-mediated chelation of residual Ca2+, enabling conditional secretion of effector proteins. By fusing the antifungal protein Bg9562 to the N-terminal secretion signal of the T3SS effector LopT and co-expressing it with the cognate chaperone SlcT, we obtained fluorescence-based evidence for T3SS-dependent delivery of Bg9562 into the hyphae of multiple Fusarium species. The engineered strain exhibited enhanced rhizosphere colonization, promoted plant growth and conferred improved protection against tomato Fusarium wilt, restoring plant height to levels approaching healthy controls. We further demonstrated the modularity of this platform by successfully transferring it into Pseudomonas koreensis D26, a strain known for its plant growth-promoting properties, indicating broad applicability across biocontrol-relevant pseudomonads. This work establishes a versatile T3SS-based delivery platform for precision biocontrol, offering a generalizable strategy for engineering beneficial rhizobacteria.
|
|
Scooped by
mhryu@live.com
June 24, 11:51 AM
|
Free-living environmental microbes at four plant-atmosphere interfaces (leaf, nectar, fruit, and bark surfaces) intercept, biotransform, and augment plant volatile signals, shaping what herbivores, pollinators, and parasitoids detect. This forum article reviews how microbes remodel these signals, the threats posed by climate change and land-use intensification, and priorities for translating microbial volatile ecology into sustainable pest management.
|

























Two pegRNAs target the genome — one at each end of the region to be replaced — generating two ssDNA flaps on the genome. Two pegRNAs target the donor plasmid — one at each end of the insert — generating two ssDNA flaps on the donor.