 Your new post is loading...

|
Scooped by
mhryu@live.com
Today, 11:33 AM
|
As for sustainable food security, plant genetic engineering has emerged as a transformative technology offering innovative solutions. This review comprehensively examines recent advances in plant genetic engineering, from technical foundations to technological innovations, and to multifaceted applications. They transcend the constraints of traditional breeding, including its long cycle and narrow genetic base, showing remarkable potential in crop improvement. By modulating key genes governing plant height, branching, leaf morphology, and root structure, plant architecture can be optimized to enhance light utilization and lodging resistance. Targeted manipulation of genes related to disease and pest resistance, and tolerance to drought, salinity, and temperature extremes, substantially improves resilience to biotic and abiotic stresses. Additionally, by fine-tuning yield determinants and by engineering photosynthetic pathways, yield potential can be effectively increased. Beyond productivity, genetic engineering facilitates nutritional fortification, improved sensory quality, and enhanced processing characteristics, paving the way for novel crop varieties that integrate nutrition with palatability. Looking forward, coordinated multi-gene editing, utilization of wild germplasm, strengthened field adaptability testing, and exploration of controllable epigenetic regulation represent key directions for the future. Collectively, these advances will drive plant genetic engineering toward greater precision, efficiency, and intelligence, providing a robust foundation for sustainable agricultural development.
|
|
Scooped by
mhryu@live.com
Today, 11:25 AM
|
Co-translational protein folding is shaped by the vectorial nature of translation, which causes residues to emerge sequentially from the ribosome. As a result, residues whose native interaction partners lie downstream in sequence cannot immediately form their native contacts and remain transiently unsatisfied until those partners are synthesized. These unsatisfied residues are vulnerable to non-native interactions and often require the engagement of co-translational chaperones. We previously developed the Native Fold Delay (NFD) metric to quantify the time lag between the synthesis of a residue and the point at which it can form all its native contacts. Here, we present the FoldDelay web server, a freely accessible platform that extends the NFD concept into a more comprehensive framework for analyzing native residue–residue contact formation during translation. Starting from user-submitted AlphaFold or PDB structures, the site identifies all N- to C-terminal residue–residue contacts, estimates their earliest possible formation times, and integrates domain annotations to distinguish between intra- and inter-domain contacts. The server provides a suite of linked interactive visualizations that allows users to explore native contact formation dynamics and detect transiently unsatisfied regions. The FoldDelay web server is freely accessible at https://folddelay.switchlab.org.
|
|
Scooped by
mhryu@live.com
Today, 2:16 AM
|
The rise of multi-drug resistant bacteria constitutes a global challenge with direct impact on millions of patients worldwide. The identification of bacterial species is essential to avoid unnecessary use of antibiotics and to minimize the emergence of additional resistance; however, there are few chemical strategies to identify resistant species in a rapid and unbiased manner. Cross-reactive arrays combine multiple sensor components to produce distinctive fingerprints for similar targets; herein, we present a cross-reactive sensing array built with long lifetime organic fluorophores. To the best of our knowledge, we synthesize the largest collection to date of bioconjugatable triangulenium fluorophores by including heteroatom and side-chain diversification for spectral and lifetime diversity, as well as water-soluble and orthogonal moieties for peptide tagging and compatibility with biological samples. After optimization, we evolve a four-fluorophore sensor array that correctly assigns all seven ESKAPEE bacterial species by combining their optical signatures. Lifetime chemical sensor arrays will open avenues for concentration-independent, reliable and sensitive optical detection without the need for prior knowledge of molecular targets. Infections caused by multi-drug resistant bacteria present a global health challenge, so the identification of bacterial species is essential to avoid the unnecessary use of antibiotics and to minimize the emergence of additional resistance. Here, the authors present a lifetime cross-reactive sensor array that correctly assigned all seven ESKAPEE bacterial species by combining their optical signatures.
|
|
Scooped by
mhryu@live.com
Today, 2:07 AM
|
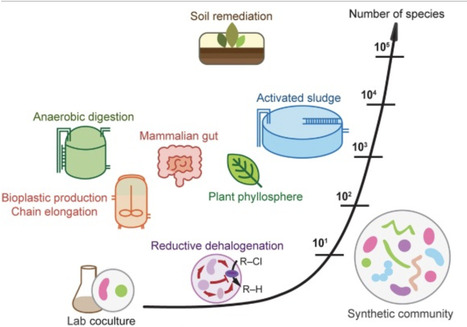
Microbial interactions hold vast potential for improving sustainability, including waste upcycling, greenhouse gas mitigation, contaminant bioremediation, and host performance. However, attempts to manage interactions in complex, open microbial communities often fall short of expectations, in part because interactions are highly context-dependent, with net effects arising from multiple component interactions, higher-order interactions, and rapid evolution—features largely revealed from studies with simplified communities. Here, we propose how these insights can be translated to complex communities by prioritizing the scales and disturbances that matter, leveraging the ecological context that constrains interactions, targeting function-critical behaviors and traits, and accounting for eco-evolutionary feedbacks. We further propose coupling top-down, trait-based, and coarse-grained approaches with bottom-up synthetic ecology approaches to diagnose failures and design robust microbial processes.
|
|
Scooped by
mhryu@live.com
Today, 2:02 AM
|
The plant immune system engages cell-surface receptors that detect microbe-associated molecular patterns to initiate pattern-triggered immunity (PTI), and intracellular receptors that sense microbe-secreted effectors to activate effector-triggered immunity (ETI). Whether additional modes of microbial detection exist remains unclear. Here, we define membrane mechanosensing immunity (MSI), a third layer of cell surface immune signaling. A bacterial lipid, the main diffusible signal factor (DSF) from Xanthomonas campestris pv. campestris, acts as a membrane-active molecule that alters plasma membrane biophysical properties, activates Mechanosensitive Channel of Small Conductance (MscS)-like (MSL)-dependent immune signaling, and triggers a broad transcriptional reprogramming that overlaps with PTI and ETI. MSI modulates PTI signaling and requires both PTI and ETI components for effective disease resistance. These findings establish the sensing of metabolite-induced membrane perturbations as a mechanism of microbial detection.
|
|
Scooped by
mhryu@live.com
Today, 1:52 AM
|
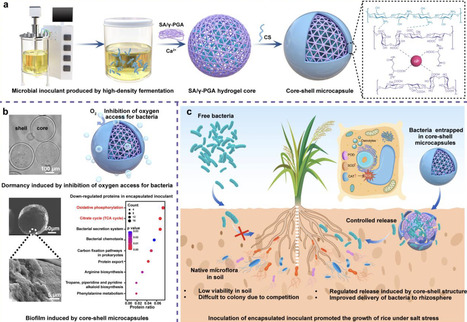
Non-spore-forming bacteria can enhance crop salinity tolerance via various strategies, but poor viability during storage and field application limits their use. Inspired by the robust structure of spore dormancy, a core-shell microcapsule consisted of sodium alginate, poly (γ-glutamic acid), and chitosan (APC) is proposed. Using Pantoea alhagi NX-11 as a model, we found that APC encapsulation significantly enhanced bacterial survival during room-temperature storage compared to free cells or alginate beads. This protective effect was further confirmed using two other non-spore-forming strains. The survival mechanism was mainly attributed to the APC-induced metabolic dormancy via suppression of the TCA cycle and oxidative phosphorylation pathways. Furthermore, inoculation with APC-encapsulated NX-11 increased dry weight of rice plant by 24.2% under salt stress in greenhouses and increased grain yield by 15.8% in saline fields, attributing to the enhanced root colonization of NX-11. Overall, this bio-inspired encapsulation strategy provides an effective approach for developing robust microbial inoculants to improve crop resilience in saline soils. Poor storage stability and soil survival limits agricultural application of non-spore forming bacteria. Here, the authors develop a hydrogel microcapsule which improves storage and soil survival, demonstrating application in improving plant salt tolerance under field conditions.
|
|
Scooped by
mhryu@live.com
Today, 1:47 AM
|
Cell–cell interactions are fundamental to multicellular organisms, governing development, maintaining tissue homeostasis, and enabling responses to perturbations. Accordingly, the development of effective tools to investigate cell–cell communication in its native context is essential for addressing fundamental biological questions. In recent years, substantial progress has been made toward studying these processes directly in vivo, where cellular dynamics, tissue architecture, and environmental cues are preserved. This review discusses these in vivo advances, focusing on how chemical and synthetic biology tools enable the interrogation and control of cell- and protein-mediated communication in living organisms. We discuss the evolution of these technologies and illustrate how they are being applied to uncover general principles of cellular interaction within complex biological systems.
|
|
Scooped by
mhryu@live.com
Today, 1:34 AM
|
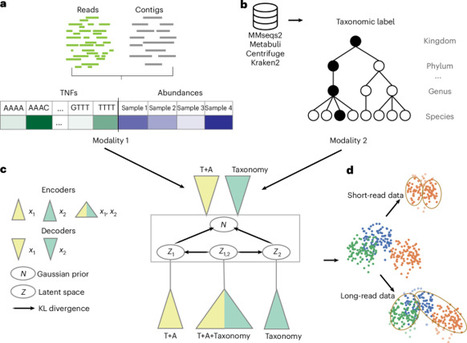
A common procedure for studying the microbiome is binning the sequenced contigs into metagenome-assembled genomes. State-of-the-art binning methods use coabundance and sequence-based motifs such as tetranucleotide frequencies, whereas taxonomic labels derived from alignment based classification have not been widely used. Here we propose TaxVAMB, a metagenome binning tool based on semisupervised bimodal variational autoencoders, combining tetranucleotide frequencies and contig coabundances with taxonomic information. TaxVAMB outperformed all other binners on CAMI2 human microbiome datasets, returning on average 29% more high-quality assemblies than the next best binner, and performed on par with the best binners on short-read datasets. On a human gut long-read dataset, TaxVAMB recovered 29% more high-quality bins. In a typical single-sample setup, TaxVAMB on average returns 83% more high-quality bins compared to VAMB. Lastly, TaxVAMB binned incomplete genomes better than any other tool, returning on average 300% more high-quality bins of incomplete genomes than the next best binner. TaxVAMB combines sequence features with taxonomic information for improved metagenome binning.
|
|
Scooped by
mhryu@live.com
Today, 1:21 AM
|
Gram-negative bacteria use a plethora of virulence factors to infect eukaryotic cells. CE-clan protease-related virulence factors were reported to act as deubiquitinases/ubiquitin-like specific proteases. Some have an additional acetyl-transferase activity. The molecular mechanisms underlying this dual activity and the physiological consequences are only marginally understood. Here, we report crystal structures for the Simkania negevensis virulence factor SnCE1 in apo-states and in complex with SUMO1. We confirm SnCE1 acting as an efficient deSUMOylase and discover an intrinsic autoacetyltransferase activity. Acetylation impairs SnCE1 tetramer formation structurally being incompatible with SUMO1 binding. We provide a model for regulation of SnCE1-mediated virulence by lysine acetylation modulating autoproteolytic processing and its subcellular distribution in the host cell. SnCE1 localizes to the endoplasmic reticulum in human cells and increases fragmentation of mitochondria. Our data provide mechanistic insights into how lysine acetylation of virulence factors is used to reprogram virulence adjusting it to the host cells’ metabolic state. Gram-negative bacteria use diverse virulence factors to infect eukaryotic cells. Here, the authors perform structure-function analyses on the S. negevensis deSUMOylase SnCE1 and provide mechanistic insights how lysine acetylation reprograms virulence adjusting it to the host cells’ metabolic state.
|
|
Scooped by
mhryu@live.com
Today, 12:59 AM
|
Since their discovery, bacteriophages—viruses that infect bacteria—have been invaluable to molecular biology and biotechnology. Renewed interest in phage-based antimicrobials, driven by the global antibiotic resistance crisis, highlights the need for improved quantitative tools. While conventional double-layer plaque assays (DLA) have provided foundational insights, they are limited by their inability to monitor infection dynamics over time and the inflexibility in experimental setups. Here, we present a high-throughput droplet microfluidics platform to quantify individual phage infection events. By co-encapsulating individual phages and bacteria in microfluidic droplets, we precisely control key experimental parameters such as exposure time and the ratio of phages to bacteria. This approach enables direct quantification of lysis events and measurement of lysis kinetics without interference from further progeny-driven infection processes inherent to bulk cultures. Applicable to diverse phage-host systems, this method offers a dynamic and accurate framework for studying phage biology and supports the development of phage-based antimicrobial strategies. Bacteriophages are key tools in molecular biology and promising agents against antibiotic-resistant bacteria. Here, the authors present a high-throughput droplet microfluidics platform that co-encapsulates single phages and bacteria to quantify individual infection events and lysis kinetics under precisely controlled conditions.
|
|
Scooped by
mhryu@live.com
Today, 12:52 AM
|
Plant-based platforms offer a sustainable alternative to chemical synthesis for producing high-value biological products, including therapeutic proteins and peptides, vaccines, antibodies, nutraceuticals, and specialised metabolites. Peptide-based therapeutics in particular hold enormous potential due to their specificity, safety, and efficacy in treating a wide range of diseases. Peptides that are comprised of the 20 canonical amino acids can be produced in plants, but challenges such as toxicity to host cells, aggregation, misfolding, susceptibility to proteolytic degradation, low expression levels, and challenges in dealing with post-translational modifications can limit successful biosynthesis. This review provides a comprehensive analysis of current successes and limitations in the field, and potential biotechnological strategies and exogenous treatments to overcome limitations, to enhance peptide production in plants.
|
|
Scooped by
mhryu@live.com
Today, 12:49 AM
|
Soil and sediment microbiomes have a central role in biogeochemical cycling, climate regulation and ecosystem resilience. However, they are increasingly degraded by land use change, pollution and climate change. Despite their foundational roles in ecosystems, these microbiomes remain under-represented in ecosystem restoration science, practice and policy. Improving the integration of microbiomes across the restoration science–practice–policy nexus is essential for achieving more effective and resilient restoration outcomes. Without this, global restoration risks neglecting the microbial foundations of functional ecosystems and long-term resilience. In this Review, we synthesize the current state of knowledge of soil and sediment microbiome restoration. We describe the major anthropogenic stressors that are degrading these microbiomes, highlighting the linked and context-dependent nature of these impacts, and evaluate existing strategies to restore them. To improve restoration effectiveness, we propose a research workflow that encompasses baseline establishment, degradation diagnostics, designing and testing interventions, research methodology selection and best practice principles. We also outline key theoretical frameworks and propose future research priorities to help soil and sediment microbiome restoration to move towards a predictive, theory-led discipline. In this Review, Wood and colleagues discuss how anthropogenic pressures are degrading soil and sediment microbiomes, propose restoration strategies and emphasize the importance of integrating microbiome science into global ecosystem restoration efforts.
|
|
Scooped by
mhryu@live.com
Today, 12:24 AM
|
The HMMER web server, available at https://www.ebi.ac.uk/Tools/hmmer, provides online access to tools from the HMMER software suite (http://hmmer.org/) for protein analysis using profile hidden Markov models. Users can perform sequence similarity searches against a range of regularly updated protein sequence databases or annotate protein sequences with domains and families using profile HMM libraries from protein family databases. Since the 2018 update, the continued exponential growth of sequence databases has necessitated substantial infrastructural improvements to maintain search performance speed and service reliability. To achieve this, the web interface has been completely reengineered using modern web technologies (JavaScript and React), providing users with an enhanced experience, including session-based search history and streamlined results visualization. The web application programming interface has been rewritten to better support programmatic access with updated endpoints and JSON-based responses. The infrastructure has been redesigned to efficiently handle searches against much larger databases through horizontal scaling and asynchronous job processing. Target database offerings have been updated to reflect current usage patterns and data availability. The HMMER web server is free and open to all users, and there is no login requirement.
|
|
|
Scooped by
mhryu@live.com
Today, 11:30 AM
|
Mycobacterial pathogens remain major global health threats, exacerbated by both rapid acquisition of antibiotic resistance and the formidable drug diffusion barrier presented by the rigid mycomembrane. These challenges have renewed interest in antimycobacterial peptides (AMyPs), a diverse class of short amphiphilic sequences capable of rapidly killing both drug-sensitive and drug-resistant mycobacteria. Beyond their intrinsic potency, AMyPs can synergize with existing antibiotics and exhibit markedly slower resistance development relative to conventional small molecules. In this review, we synthesize recent advances spanning natural bioprospecting, mechanism-guided rational design, and chemical optimization strategies that have yielded increasingly potent and selective AMyP candidates. We further highlight the rapid emergence of artificial intelligence–driven discovery platforms, which leverage machine-learning models trained on curated, mycobacteria-specific data sets to predict and refine novel AMyPs with growing accuracy. Together, these technologic, biologic, and computational advances outline a rapidly expanding landscape for AMyP-based therapeutic development and establish a foundation for next-generation antimycobacterial drug design. amp
|
|
Scooped by
mhryu@live.com
Today, 2:33 AM
|
Here, we report selective covalent assembly of Gram-negative bacteria and synthetic polymers into functional living materials. We discovered that triblock polymers decorated with vinyl sulfone (VS), a motif that forms a stable covalent bond with cysteine residues on surface proteins, yielded stable covalent assembly with bacterial cells. Notably, we found that these assemblies were uniquely cell-type-specific, occurring only in Gram-negative bacteria, highlighting differences in surface structures and the macromolecular diffusion barrier across bacterial species. We also demonstrated that assembling engineered cells into materials results in situ melanin production from living materials, with robust biocontainment and mechanical reinforcement. Spontaneous enrichment of tyrosine-derived red pigments in the supernatant showcases the effect of confinement in a complex biochemical pathway. This work establishes a platform for encoding complex, engineered, and evolved functions of Gram-negative bacteria into synthetic materials, enabling the development of a wide range of material-based bioreactors.
|
|
Scooped by
mhryu@live.com
Today, 2:11 AM
|
Uncovering what drives select biomolecules to form phase-separated condensates in vivo and identifying their physiological significance are topics of fundamental importance. Here, we show that nitrogen-starved E. coli produces long-chain polyphosphates, which scaffold the RNA chaperone Hfq into high molecular weight complexes, which eventually phase separate together with components of the RNA translation and processing machinery. The presence of polyphosphate within these condensates controls Hfq function by selectively stabilizing polyadenylated RNAs involved in transcription and protein translation and by promoting interactions with translation- and RNA-metabolism-associated proteins involved in de novo protein synthesis. Lack of polyphosphate significantly impairs condensate formation, increases cell death, and hinders recovery from N-starvation. In functional analogy, we demonstrate that polyP contributes specifically to the formation of Processing (P)-bodies in human cell lines, revealing that a single, highly conserved and ancestral polyanion serves as a modulator for functional phase-separated condensate formation across the tree of life.
|
|
Scooped by
mhryu@live.com
Today, 2:05 AM
|
Competition for a single limiting resource is expected to lead to competitive exclusion, yet diverse microbial communities persist even in nutrient-poor environments. Cross-feeding of essential metabolites is one mechanism that can promote coexistence between species, but its contribution is difficult to pinpoint experimentally. Here, we studied a prototroph-auxotroph pair growing on a single carbon source in chemostats. In minimal medium, the prototroph Comamonas testosteroni (Ct) supplied thiamine to the thiamine-auxotroph Ochrobactrum anthropi (Oa), allowing stable coexistence in agreement with consumer-resource theory. Contrary to our expectation that supplying thiamine would remove the dependency and lead to exclusion of Ct, coexistence persisted even when thiamine was supplemented. Our theoretical analysis showed that coexistence between competitors can be maintained by trace concentrations of an additional metabolite if it is taken up at sufficiently high affinity by the weaker competitor. Consistent with this prediction, targeted metabolomics and spent-medium assays identified growth-enhancing compounds at micromolar concentrations in Oa spent medium and as residues in fresh medium. Model analysis further showed that such weak positive effects can qualitatively change coexistence outcomes in chemostats while remaining undetected in standard batch interaction assays. Together, our results show that trace metabolites and subtle positive effects can reshape coexistence outcomes and should be incorporated into ecological models and interaction measurements.
|
|
Scooped by
mhryu@live.com
Today, 1:58 AM
|
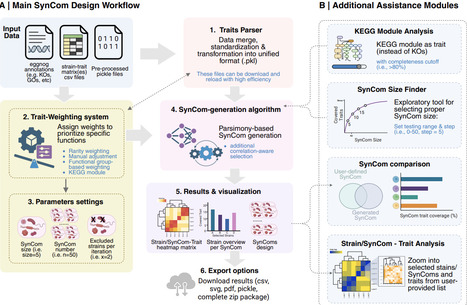
Synthetic microbial communities (SynCom) are essential tools for dissecting the causal mechanisms in host-microbiota interactions. To date, however, SynCom design suffers from a lack of standardization, typically oscillating between arbitrary strain selection and computational pipelines that misalign with experimental design. As microbiome research transitions toward functionally defined community systems with reproducible experimental outcomes, there is a strong need for a user-friendly platform that integrates multi-dimensional genomic and/or biological data into a standardized and tailored SynComs design. Here, we present SynCom101, a web-based platform that democratizes the design of reproducible, hypothesis-driven SynComs. SynCom101 accommodates diverse input formats including genomic annotations and laboratory-obtained phenotypic traits, allowing users to customize their design criteria with high flexibility. The platform utilizes a parsimony algorithm to ensure computational scalability for large datasets, complemented by an optional correlation-aware mode to account for microbial compatibility and co-occurrence patterns when ecological interactions among strains are available. A core innovation of SynCom101 is its suite of trait-weighting modules, which empowers researchers to strategically guide the selection algorithm toward maximal functional trait coverage, the emulation of natural community architectures, or the enrichment of positively correlated microbial assemblages to enhance community stability. We showcase the functionalities of the platform by in silico design of communities from different datasets, demonstrating its capacity to generate concise, functionally prioritized SynComs aligned with targeted design objectives. By providing a transparent, parameter-documented workflow, SynCom101 ensures that community design is no longer a "black box" but a reproducible scientific record. This platform establishes a necessary standard for in silico community assembly, facilitating the transition from descriptive microbiome studies toward high-throughput, predictive functional screening and cross-study comparability. https://syncom101.bioinformatics.nl/ The datasets used for case studies are available on Zenodo (https://doi.org/10.5281/zenodo.18310451). The source code is available at Git (https://git.wur.nl/jiayi.jing/syncom101).
|
|
Scooped by
mhryu@live.com
Today, 1:48 AM
|
Microalgae are increasingly recognized as versatile biological platforms capable of supporting sustainable food, energy, environmental, and industrial systems. This review critically evaluates recent advances in microalgal biotechnology, with a focus on applications in which biological performance, resource recovery, and scalability intersect. Emphasis is placed on microalgal biomass composition, including proteins, lipids, polyunsaturated fatty acids, pigments, and polysaccharides, and how these components underpin applications in food, feed, biofuels, bioremediation, and bioproduct development. Environmental applications, particularly wastewater treatment and carbon capture, are examined as integrated systems that couple nutrient removal with biomass valorization. Advances in strain improvement, including genetic engineering and multi-omics approaches, are discussed alongside persistent limitations related to species-specific transformability, production costs, and downstream processing. Rather than providing an exhaustive survey, this review highlights cross-sectoral trends, quantitative performance metrics, and technological bottlenecks that constrain commercialization. By comparing biological potential with engineering and economic realities, this work identifies priority research directions needed to advance microalgae-based systems within circular bioeconomy frameworks.
|
|
Scooped by
mhryu@live.com
Today, 1:44 AM
|
Bacteria encode diverse defence systems, including restriction–modification and CRISPR–Cas, that cleave nucleic acid to protect against phage infection. Bioinformatic analyses demonstrate that many recently identified antiphage defence operons comprise a nuclease and NTPase protein, suggesting that additional nucleic acid-targeting systems remain to be understood. Here we develop large-scale comparative cell biology and biochemical approaches to analyse 16 nuclease–NTPase systems and define molecular features that control antiphage defence. Purification, biochemical characterization and in vitro reconstitution of nucleic acid degradation demonstrates that protein–protein complex formation is a shared feature of multigene nuclease–NTPase systems. We show that PaAbpAB, BtHachiman and EcPD-T4-8 system nucleases use highly degenerate recognition site preferences to enable broad nucleic acid degradation, and the Azaca system exhibits specific phage targeting through the recognition of modified phage genomic DNA. Our results uncover principles of antiphage defence system function and highlight the mechanistic diversity of nuclease–NTPase systems in bacterial immunity. A large-scale, comparative cell biology and biochemical screen defines the molecular features controlling antiphage defence systems that encode nuclease effectors and accessory NTPase proteins.
|
|
Scooped by
mhryu@live.com
Today, 1:31 AM
|
Dissecting the functional impact of genetic mutations is essential to advancing our understanding of genotype-phenotype relationships and identifying therapeutic targets. Despite progress in sequencing and genome editing technologies, proteome-wide mutation effect prediction remains challenging. Here we show that evolutionary information alone enables accurate prediction of mutation effects across entire proteomes. ProteoCast is a scalable and interpretable computational method that leverages protein sequence conservation to classify genetic variants and identify functionally important protein sites. We apply ProteoCast to the complete Drosophila melanogaster proteome (22,000 isoforms, 300 million mutations) and validate it against nearly 400,000 natural and experimental variants. It correctly classifies 85% of known lethal mutations as functionally impactful versus 13-18% of population variants. ProteoCast-guided genome editing experiments confirm these predictions. Moreover, ProteoCast successfully identifies functionally important protein modification sites and binding motifs. ProteoCast provides a publicly available resource and deployable pipeline for studying gene function and mutations in any organism. This study introduces ProteoCast, which uses protein sequence conservation to predict mutation effects proteome-wide. Validated on 400,000 Drosophila variants and genome editing experiments, it accurately identifies functionally critical sites in any organism.
|
|
Scooped by
mhryu@live.com
Today, 1:06 AM
|
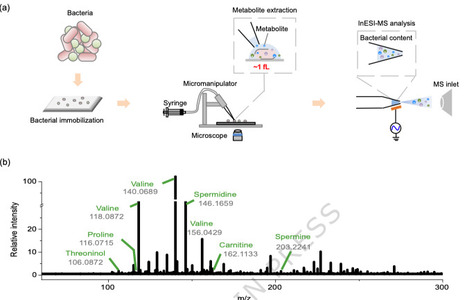
Bacterial metabolism is a complex system of interwoven pathways coordinated by an intricate, multilayered regulatory network. Dissecting the metabolism at single bacterial level is highly challenging. Herein, we greatly increase the sensitivity of the bacterial measurement with our home-made single-bacterium metabolome (SinBactM) mass spectrometry platform, which combines micro-extraction with induced nanoelectrospray ionization mass spectrometry, for mapping metabolites of the primary bacterium. The universal applicability of the SinBactM platform is demonstrated by successfully discriminating distinct metabolic profiles across different bacterial species. By measuring the bacterial uptake of antibiotics and observing the dynamic metabolic changes in response to external stimuli, the accuracy and reliability of SinBactM platform are further validated. We apply SinBactM to characterize clinical heteroresistant K. pneumoniae strains metabolic alteration under antibiotic perturbation. Several different subpopulations resulting from antibiotic perturbation are classified based on their metabolome, while pseudotemporal ordering reveals a trajectory of metabolic change across subpopulations. Thus, SinBactM platform paves the way for profiling metabolomics of individual bacterial cells, which might provide an exciting dimension to a thorough understanding of antibiotic-resistant evolution for in-depth interrogation of resistant mechanisms. Here the authors present a single-bacterium metabolomics platform that enables sensitive metabolic profiling of individual bacteria, revealing metabolic heterogeneity in heteroresistant K. pneumoniae strains and providing insights into bacterial survival and resistance.
|
|
Scooped by
mhryu@live.com
Today, 12:54 AM
|
Human bacterial pathogens can be part of the soil microbiota, but their prevalence and global distribution is unknown. Using metagenomic approaches, Xiong and colleagues identified 80 bacterial taxa of human pathogens in 1,602 soil samples collected across all continents. Among these, 25 taxa, including highly pathogenic ones, were present in high relative abundance in more than 80% of all soil metagenomes. The relative abundance of these dominant pathogens correlated positively with the presence of pathogenic features such as virulence factors in the metagenomic samples. Moreover, these distributions correlated with global mortality patterns of the associated infectious diseases, which suggests that soil-associated human pathogens represent a potential global risk. These dominant pathogens were especially prevalent in tropical and temperate ecosystems, their distribution correlating positively with rainfall and temperature but negatively with soil microbiota diversity. Predictions taking into account trends of global precipitation and temperature changes revealed a global increase in the proportion of human pathogens in the soil worldwide. These results suggest that knowledge about the prevalence of human bacterial pathogens in soils, influenced by climatic conditions and soil biodiversity, is important to improve risk assessment and management strategies against infectious diseases.
|
|
Scooped by
mhryu@live.com
Today, 12:50 AM
|
CRISPR–Cas12a is a versatile RNA-guided nuclease that has rapidly gained prominence for its dual functionality in genome editing and nucleic acid detection. In this Review, we discuss the structural, biochemical and mechanistic features of Cas12a that underpin its autonomous processing of the guide RNA and indiscriminate cleavage of single-stranded DNA, which enable Cas12a applications ranging from gene therapy to rapid diagnostics. We discuss key allosteric regulators and functional modules that orchestrate Cas12a activity, focusing on the core regulatory structural elements that control maturation of the guide RNA, target specificity, and both cis-cleavage and trans-cleavage activities, including the determinants of off-target cleavage. We provide a comparative analysis of Cas12a and the widely used Cas9, which further illuminates the distinctive attributes of Cas12a, and discuss recent advances in the characterization of its orthologues and in the development of engineered variants that expand its capabilities. Collectively, we present a comprehensive understanding of Cas12a and its increasing impact on biotechnology, therapeutics and molecular diagnostics. This Review discusses the unique features of the functionally versatile nuclease Cas12a, including allosteric regulation, structural elements controlling guide RNA maturation, target specificity, cis-cleavage and trans-cleavage activities and determinants of off-target cleavage. These features increase the applicability of Cas12a in biotechnology, therapeutics and molecular diagnostics
|
|
Scooped by
mhryu@live.com
Today, 12:37 AM
|
Microbial electrochemical technologies connect living microorganisms with electrical systems to enable sustainable processes. These systems use the natural electron transfer abilities of microbes to support clean energy generation, transform waste and pollutants, and recover valuable resources. Microorganisms act as living catalysts whose biological traits and energy requirements determine system performance. Recent advances have expanded these technologies toward chemical production and new electronic applications, while improved engineering has increased their readiness for practical use. At the same time, these systems offer a powerful platform for studying fundamental microbial processes in real time. This review outlines the biological foundations of microbe–electrode interactions, highlights current applications relevant to circular resource use, and identifies scientific and engineering challenges that must be addressed to unlock broader societal and economic benefits. The goal is to emphasize the growing potential of microbe–electrical interfaces to support transformative solutions for environmental sustainability. Microbial electrochemical technologies can enable clean energy, waste conversion, and resource recovery, according to a literature synthesis on microbe-electrode interactions and engineered bioelectrochemical systems.
|


























encapsulation