Your new post is loading...

|
Scooped by
mhryu@live.com
Today, 5:04 PM
|
Bacterial genomes represent dynamic ecological systems in which highly dynamic accessory genome element compositions drive evolution. Emerging evidence suggests that bacterial defence systems, which protect against phages and other genetic elements, can interact cooperatively, competitively, and antagonistically to influence horizontal gene transfer, shape phage susceptibility, and diversify genomes across environments. Recent ecological studies reveal non-random co-occurrence and avoidance patterns among defence systems suggesting that these patterns may emerge from ecological and evolutionary interactions rather than chance. Hence, these patterns need exploring in the context of ecological niche and co-localization to identify putative functional compatibilities and elucidate how defence systems shape the accessory genome. To characterize these patterns, we analyzed the distributions of defense systems and other accessory genome elements in a curated global dataset of 2940 Pseudomonas aeruginosa. Defence system content varied by ecological niche, with higher numbers in non-cystic fibrosis derived isolates (average n = 7.9) compared to cystic fibrosis-derived isolates (average n = 6.5). There were also multiple associations (n = 426) and dissociations (n = 50) among defence systems, and among other accessory genome elements, many with a plausible biological explanation. We also found that defence and anti-defence systems engage in more interactions than other accessory genome element types (e.g. antimicrobial resistance genes, plasmids) suggesting that they are a major driving force in the ecological dynamics of bacterial genomes. These patterns provide new insights into the evolutionary forces shaping bacteria, and provide a valuable resource of robustly quantitated interactions, establishing a baseline for future mechanistic and ecological investigations of defence system interactions.
|
|
Scooped by
mhryu@live.com
Today, 3:41 PM
|
This study introduces BioLumiPi, a novel, low-cost, and non-invasive biosensor plant, engineered by combining a phosphate (Pi) deficiency-induced promoter with a fungal bioluminescence system in tobacco. BioLumiPi plants emit light that accurately reflects endogenous Pi deficiency and responds specifically to Pi levels. Coupled with deep learning analysis, this technology offers a proof-of-concept strategy for real-time visualisation and prediction of nutrient stress to optimise fertiliser application and advance sustainable agriculture.z
|
|
Scooped by
mhryu@live.com
Today, 3:21 PM
|
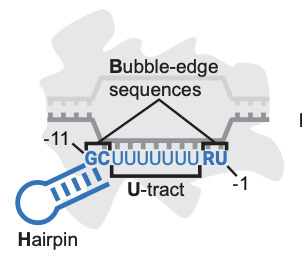
Transcription terminators are universal landmarks that delimit RNAs and tune downstream gene expression, yet the sequence rules that define them remain elusive. The classical model of bacterial intrinsic termination - an RNA hairpin followed by a uracil-rich tract - is incomplete, as stem-preserving mutations can abolish termination. By precisely mapping termination sites across 10^4 sequences, we uncovered a previously unresolved feature required for intrinsic termination: dinucleotides positioned at both edges of the transcription bubble, resembling the elemental RNA polymerase pause signal. Together, hairpin, U-tract and bubble-edge sequences (HUB) account for most variation in termination efficiency and pinpoint bona fide terminators across diverse bacterial phyla. These findings establish HUB as the defining element of intrinsic terminators and provide a framework for decoding and engineering gene expression across genomes.
|
|
Scooped by
mhryu@live.com
Today, 3:08 PM
|
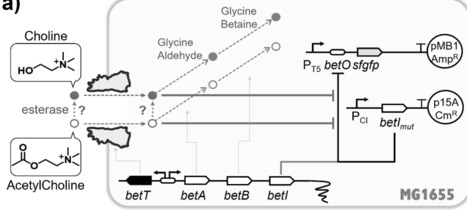
Acetylcholine (ACh) is widely recognized as a neurotransmitter in animals; however, its potential relevance to microbial physiology remains largely unexplored. Here, we report that the BetI regulatory system of E. coli, previously characterized as a choline-responsive osmoprotective pathway, exhibits a measurable transcriptional response to extracellular ACh. Using a recombinant reporter strain, we demonstrate that ACh induces BetI-dependent gene expression and that the shape and sensitivity of the response curve are markedly altered by genetic perturbations of pathway components, including BetT overexpression and BetA deletion. While the underlying molecular mechanism remains unresolved, these results establish that E. coli can functionally respond to ACh at the whole-cell level. Given that ACh is readily hydrolyzed into metabolites relevant to microbial metabolism and is supplied in animal tissues from sources beyond neurons, our findings suggest that acetylcholine responsiveness may be more widespread among host-associated microorganisms than previously appreciated.
|
|
Scooped by
mhryu@live.com
Today, 2:47 PM
|
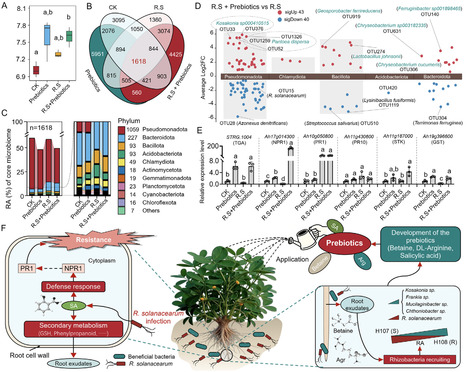
Bacterial wilt caused by Ralstonia solanacearum compromises the yield and quality of peanut (Arachis hypogaea L.). While rhizosphere microbiome-assisted defense is known, how resistant plant genotypes orchestrate this process remains unclear. Here, we integrate multi-omics analyses of resistant and susceptible peanut genotypes to uncover a genotype-specific defense mechanism. The resistant genotype selectively recruits beneficial bacteria (e.g., Kosakonia and Frankia), which coincides with activated salicylic acid (SA)-dependent systemic acquired resistance (SAR). Crucially, we identify keystone rhizosphere metabolites (including betaine, arginine, and SA) that are positively correlated with both beneficial microbiome assembly and SAR gene expression, establishing a self-reinforcing defense loop. Leveraging these insights, we develop a prebiotic formulation that enhances beneficial microbial recruitment and stimulates SAR. Field trials demonstrate that the prebiotics reduce bacterial wilt incidence from 84.2% to 5.0% and increase yield by 12.9%–20.3%. Collectively, our study reveals a synergistic microbiome-immune co-regulation mechanism in peanut and delivers a translatable solution for sustainable disease management.
|
|
Scooped by
mhryu@live.com
Today, 10:59 AM
|
Engineered therapeutic microbes for intestinal inflammation must be capable of gut colonization, sensitive detection of disease-associated biomarkers and targeted delivery of therapeutic molecules. Examples of microbes displaying all three characteristics are limited. Here we engineered Bacteroides thetaiotaomicron, a human gut commensal bacterium with colonization ability, colonic tropism and innate anti-inflammatory properties, as a chassis to create programmable bacterial strains termed Btbots. We developed genetic circuits that were integrated into the bacterial chromosome to sense two intestinal inflammation biomarkers, deoxycholic acid (DCA) and nitric oxide (NO), and to enhance surface display and secretion of therapeutic molecules in response. Btbots with these biosensors were developed that either display trefoil factor-3 to facilitate mucosal repair or secrete interleukin-35 to suppress inflammation. These Btbots sensed DCA and NO biomarkers and released therapeutic agents, alleviating colitis and modulating the gut microenvironment and microbiota in mouse models. This work establishes a proof of concept for localized sensing and consequent therapeutic molecule release for gastrointestinal applications, whose clinical potential awaits further investigation. Engineered Bacteroides thetaiotaomicron, Btbots, use a genetic AND circuit to sense nitric oxide and deoxycholic acid as inflammatory signal in the gut to release therapeutic molecules, TFF3 and IL-35, that alleviate colitis in mice.
|
|
Scooped by
mhryu@live.com
Today, 10:22 AM
|
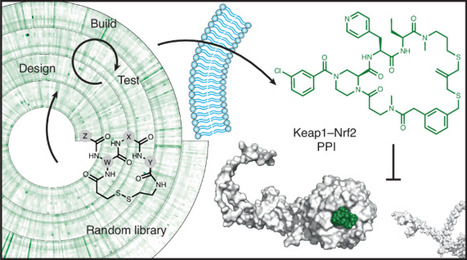
Small, nonpolar cyclic peptides can both bind challenging targets and cross cell membranes, making them attractive for addressing currently undruggable targets such as many protein–protein interactions (PPIs). However, developing such compounds de novo without prior information about lead structures such as natural ligands or fragments remains a notable challenge. Here we show that functional screening of structurally highly diverse cyclic peptide libraries synthesized at nanomole scale allows identification of sub-kDa inhibitors of a PPI. By screening 15,360 fully random cyclic peptides, we were able to identify an inhibitor of the E3 ligase adaptor Keap1 and its substrate Nrf2. Optimization by rapid design–build–test cycles produced a membrane-permeable compound active in live cells. This study demonstrates that large, diverse cyclic peptide libraries can enable the discovery of cell-permeable PPI inhibitors from the ground up, providing a way to harness the powerful modality of small cyclic peptides to address often difficult-to-target intracellular interactions. Membrane-permeable cyclic peptides offer access to difficult intracellular targets but discovery remains challenging. Here the authors show that cell-active cyclic peptides can be identified by screening sufficiently large and diverse libraries of small synthetic peptides.
|
|
Scooped by
mhryu@live.com
May 31, 9:57 AM
|
Bacteria are able to coordinate cell growth and genome replication in different growth conditions.The DNA-binding protein DnaA is responsible for determining initiation of replication, thereby playing a central role in this coordination. Theoretical and experimental studies have shown that stability of the cell cycle requires an ultrasensitive response, i.e., a sharp dependence of the initiation firing rate on the cell volume. However, the source of such ultrasensitivity remains elusive. In this work, we elucidate how the structure and binding affinities of the DnaA regulatory system determine its ultrasensitive response. Our theory sets precise constraints on inding parameters, that are necessary for cell cycle stability. Our findings show how the variety of regulatory mechanisms of the DnaA system are required for ultrasensitivity across growing conditions.
|
|
Scooped by
mhryu@live.com
May 31, 9:40 AM
|
We evaluate whether tryptophan (W), widely thought to be the last of the 20 canonical amino acids added to the genetic code, was already present in the Last Universal Common Ancestor (LUCA). We reconstruct the evolutionary history of tryptophanyl-tRNA synthetase (WRS), the enzyme that attaches W to its tRNA, and the related tyrosyl-tRNA synthetase (YRS). We identify and exclude sequences derived from ancient recombination between archaeal and bacterial YRSs. Diverse rooting methods, including a novel approach exploiting time non-reversible evolution, all place the root between bacterial and archaeal YRS rather than between YRS and WRS. This supports post-LUCA WRS origination in Archaea, followed by its horizontal transfer to Bacteria. However, ancestral sequence reconstruction suggests that Archaea were depleted for W while Bacteria were not, and enzymes essential for W biosynthesis emerged in Bacteria. This suggests that W usage originated in Bacteria, with later WRS emergence in Archaea allowing the archaeal genetic code to converge with the bacterial code. The universality of the genetic code is usually attributed to common descent from LUCA, but the final step making the code universal was instead achieved by horizontal gene transfer. This gives credence to similar mechanisms for earlier steps in genetic code evolution.
|
|
Scooped by
mhryu@live.com
May 31, 1:08 AM
|
As mobile genetic elements, transposons play a crucial role in the adaptive evolution and genome engineering of industrial microorganisms. Their applications range from high-throughput functional genomics, enabling systematic genotype-phenotype mapping via transposon sequencing, to the construction of random integration libraries for chassis development and directed evolution. Despite the emergence of precise editing tools such as CRISPR-Cas, transposon technology remains indispensable in non-model industrial strains owing to its operational simplicity and high efficiency. Recent discoveries of novel transposon systems, along with their functional enhancement, have further expanded their utility. Looking ahead, integrating transposon technology with strategies such as AI-assisted design and CRISPR-Cas-based systems will greatly advance our ability to decipher and engineer industrial microbial cell factories. This review summarizes the principles, challenges, and opportunities of transposon-associated technologies in industrial microorganisms, offering new insights into their roles in industrial biotechnology.
|
|
Scooped by
mhryu@live.com
May 31, 1:04 AM
|
Our current industrial, agricultural, and medical practices exploit the extraordinary biodiversity generated through billions of years of natural evolution. Despite their high fitness in native habitats, biomolecules and organisms are often not optimally suited for industrial and medical use. Synthetic evolution leverages technologies such as DNA synthesis, CRISPR–Cas engineering, and synthetic biology to enable continuous in vivo mutagenesis of biomolecules or organisms to improve specific desirable characteristics. This review presents the latest mutagenesis toolkits classified by mutational scale: genome-wide, medium-scale, and site-specific, each tailored to different application scenarios. We discuss the mechanisms and capabilities underlying each scale, analyze current limitations, and highlight the untapped potential of next-generation gene-editing technologies, high-throughput screening, and artificial intelligence in advancing synthetic evolution.
|
|
Scooped by
mhryu@live.com
May 31, 12:42 AM
|
Programmable control of microbial gene expression by plant hosts could enable a new generation of precision agricultural biotechnology. Here, using O-methyl-L-tyrosine (OMY) as a model compound, we establish non-standard amino acids (nsAA) as a platform for plant-based control of associated microbial activity. We use genetic code expansion to engineer OMY-dependent control of protein synthesis in the soil bacterium Bacillus subtilis. Then, we engineer agronomically diverse plants, including Arabidopsis, tomato and poplar, to biosynthesize OMY. We show that plant-derived OMY can stimulate gene expression in both model and wild soil bacteria and demonstrate how inducible and tissue-specific expression of a single biosynthetic enzyme by the plant enables tight, on-demand control over microbial activity. This work establishes nsAAs as a tool for programming plant-microbe partnerships.
|
|
Scooped by
mhryu@live.com
May 31, 12:35 AM
|
Cyclic di-GMP (c-di-GMP) is a key bacterial second messenger that regulates a wide range of cellular processes, including biofilm formation and virulence. Multi-domain one-component systems regulate c-di-GMP synthesis and turnover in response to external signals. Periplasmic sensing of environmental cues is performed by versatile but conserved sensory domains, including members of the CHASE4 superfamily. Here, we explore the promiscuity of the CHASE4 domain in c-di-GMP signal transduction in Pseudomonas aeruginosa by analyzing two CHASE4-containing transducers from the virulent PA14 strain, namely PA14_53310 and PA14_37690. With the integration of biochemical and biophysical methods, such as UV-Vis spectroscopy, circular dichroism, and isothermal titration calorimetry, we demonstrate that the CHASE4 domains of these proteins possess different ligand specificities. We show that PA14_53310 is a diguanylate cyclase, and that its enzyme activity is controlled by heme binding to its periplasmic CHASE4 domain. The PA14_37690 CHASE4 domain, on the other hand, does not bind heme, but likely recognizes copper, indicating a role in metal ion sensing. A comparison with the PAO1 counterpart, that is, PA0847 and PA2072, is discussed and the divergences highlighted. This work demonstrates that the CHASE4 domain is a multifunctional sensory module that can be calibrated to detect a range of environmental cues, thus providing a mechanism for c-di-GMP signaling refinement in P. aeruginosa. These results shed light on the molecular basis for the functional diversification of CHASE4-containing one-component systems and their role in bacterial adaptation.
|
|
|
Scooped by
mhryu@live.com
Today, 5:02 PM
|
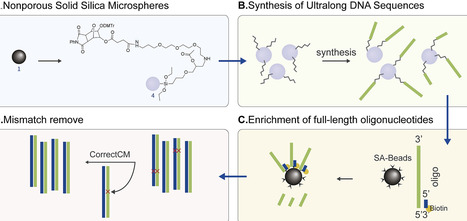
Synthetic biology and advanced genetic engineering applications rely heavily on the efficient construction of large and complex DNA sequences. Current DNA synthesis technologies have limited capacity to efficiently generate ultralong oligonucleotides for complex gene construction, particularly those with extensive repetitive motifs and uneven base distribution. Here, we report a novel platform named UCOS (Ultralong Complex Oligonucleotides Synthesis) that enables the efficient synthesis of long, complex, and challenging DNA fragments. This platform employs nonporous silica microspheres as the solid support instead of the traditional controlled pore glass solid support, full-length enrichment based on 5′ flank sequence hybridization and an error-removing enzyme for correct sequence selection, substantially enhancing the fidelity of intricate, ultralong oligonucleotides. Using this approach, we successfully synthesized challenging sequences ≤600 nt in length, encompassing tandem repeats and uneven base distributions. Overall, this novel platform demonstrates exceptional efficiency and reliability in handling ultralong DNA fragments with highly repetitive and complex features. This novel platform provides a strong foundation for advancing synthetic biology and metabolic engineering, showing great potential as a powerful tool for constructing challenging genes and enabling the customized synthesis of functional genetic elements for complex genetic programmes and synthetic genomics.
|
|
Scooped by
mhryu@live.com
Today, 3:33 PM
|
Racemisation has traditionally been regarded as an undesirable side reaction that must be minimised or eliminated during chemical synthesis. However, racemisation is also a naturally occurring process that amino acids undergo as part of normal biological and evolutionary dynamics. Increasing evidence suggests that this stereochemical conversion plays important roles in biological systems and has been used to investigate cellular growth, ageing and protein turnover. While maintaining stereochemical purity is essential during synthetic processes, often achieved through milder coupling conditions and controlled reaction environments, it is important to recognize that racemization can occur even under relatively mild, physiological conditions, without the need for elevated temperatures or extreme pH values. This review examines racemisation from both synthetic and biological perspectives, together with its biological applications. Particular attention is given to spontaneous racemisation as a contributing factor in ageing, where disruption of chiral balance and the accumulation of irreversible post-translational modifications (PTMs) are frequently observed. We discuss the mechanisms and factors that trigger this stereochemical conversion, strategies for minimising it during synthesis, and the potential benefits of understanding and exploiting racemisation. Applications such as age estimation in forensic science and insights into neuronal development are also highlighted.
|
|
Scooped by
mhryu@live.com
Today, 3:12 PM
|
Biological macromolecules, such as proteins, are made of concatenated building blocks. We hypothesized that individual protein residues could be imaged by anchoring their side chains to a swellable polymer, cleaving backbone amide bonds, and expanding residues away from each other to a degree that enables them to be visualized separately. We introduce thousandfold expansion microscopy (1000ExM), a four-network interpenetrating hydrogel architecture that enables successive expansion from ~18-fold to >1000-fold (one billion-fold in volume). Protein and peptide structures are maintained across these expansion factors, as verified by analyses of proteins with known structures (nanobodies, GFP) and a well-studied peptide (mCLING). Computational analysis indicates that 1000ExM resolves adjacent amino acid residues, thereby achieving sub-nanometer precision on conventional light microscopes. We anticipate that 1000ExM will find wide utility in protein visualization and identification, potentially even in intact cells and tissues.
|
|
Scooped by
mhryu@live.com
Today, 2:53 PM
|
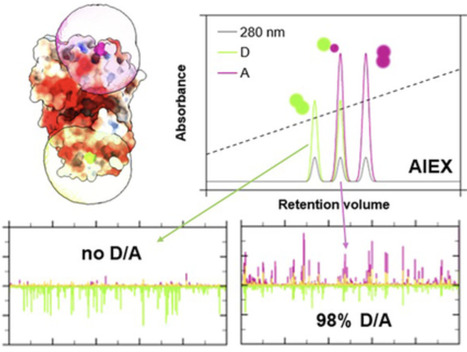
Förster resonance energy transfer (FRET), an optical distance ruler, provides the unique ability to monitor molecular interactions and conformational changes in single biomacromolecules and multi-subunit complexes. It has proven to be a powerful tool to study enzymatic reactions, membrane transport, protein folding, but also the properties of nucleic acids, proteins, molecular motors, and many other biological systems and processes. A prerequisite for FRET is targeted (covalent) labeling of macromolecules with two distinct fluorophores. Here, we present a strategy for stochastic labeling of protein residues with the required donor and acceptor dyes via anion exchange chromatography. While this technique has been used before for this purpose, we provide a conceptual basis to systematically design a purification protocol for an arbitrary choice of fluorophores. By characterizing the interaction of hydrolyzed fluorophore-maleimides with the column material, we are able to select (and predict) which pairs of fluorophores allow a successful purification of donor–acceptor-labeled protein with yields up to 98%. We demonstrate the capabilities of the method for bulk and single-molecule FRET assays of various bacterial substrate-binding proteins, which are examples of soluble, folded, and well-characterized protein systems.
|
|
Scooped by
mhryu@live.com
Today, 2:10 PM
|
Transcriptional terminators are key to defining transcript boundaries, ensuring mRNA maturation, and maintaining expression stability, yet they remain underexplored in filamentous fungi. Here, we systematically benchmarked 15 heterologous 3′ terminators in Aspergillus oryzae by measuring protein output via mCherry fluorescence and transcriptional readthrough based on a quantitative readthrough index (RTI) derived from RT-qPCR. Across the tested terminators, mCherry expression varied more than 5-fold and RTI spanned over 50-fold, indicating substantial functional diversity. Expression strength and termination efficiency were only weakly correlated (Spearman ρ = −0.39, p = 0.165), indicating that these two properties are largely independent and can be optimized separately. Strong terminators -TactA, Txyn1, TmutA and TtrpC combined good mCherry expression (>ΔT) and small readthrough (RT < 0.15). In contrast, short synthetic sequences (≤70 bp) showed low mCherry output accompanied by transcriptional leakage. Interestingly, widely used toolkit elements such as Ttef1 and Tcyc1 performed poorly in A. oryzae, highlighting the importance of empirical characterization over assumed transferability. Polyadenylation site mapping by 3′ rapid amplification of cDNA ends (3′RACE) reveals high-performing terminators that possess focused cleavage sites, continuous poly(A) tails, and recognizable A[AT]TAAA-like motifs with moderately AU-rich upstream regions, whereas weak terminators exhibited dispersed cleavage and sparse upstream sequence elements. Targeted disruption of the canonical AATAAA hexamer in TactA did not significantly impair expression or termination efficiency, demonstrating that the canonical PAS is not strictly required when AU-rich upstream sequence elements are present. Terminator performance rankings were robust across three carbon sources and were confirmed using a secreted NanoLuc luciferase reporter, demonstrating that functional behavior is largely sequence intrinsic. These findings demonstrate a functional terminator architecture and suggest a practical guideline for terminator selection in A. oryzae.
|
|
Scooped by
mhryu@live.com
Today, 10:30 AM
|
Accurately predicting protein-protein interactions (PPIs) in dimeric complexes remains a fundamental challenge in computational biology. Although existing PPIs prediction models, such as AlphaFold-Multimer (AF-Multimer) and AlphaFold3 (AF3), have achieved impressive performance, they still suffer from unsatisfactory accuracy due to the limited availability of protein dimer structures, whose collection is both expensive and labor-intensive. Here, we introduce a simple yet effective pre-training method, termed split and merge proxy (SMP), that leverages abundant monomeric proteins to simulate various PPIs tasks for the first time. Specifically, SMP constructs pseudo-dimers by splitting monomer data into two subunits, referred to as pseudo-receptors and pseudo-ligands, and trains models to merge them back by predicting their pseudo interactions (e.g., contact or docking). This proxy task enables large-scale pre-training without additional cost. Models pre-trained with SMP and subsequently fine-tuned on real protein dimer datasets demonstrate consistently improved accuracy and generalization across multiple benchmarks, surpassing strong baselines. Notably, SMP delivers more accurate structure predictions than both AF-Multimer and AF3 on several CASP15 dimer targets. Our findings highlight SMP as a scalable strategy for harnessing monomeric data to advance protein complex modeling, providing insights into the linkage between monomers and multimers. Accurate prediction of protein-protein interactions is limited by the scarcity of high-quality complex structures. Here, authors introduce SMP, a strategy that leverages pseudo-dimers derived from monomers to improve accuracy and generalization across diverse protein interaction applications.
|
|
Scooped by
mhryu@live.com
May 31, 3:53 PM
|
Non‑alcoholic beers (NABs) are a rapidly expanding market. However, producing high‑quality products remains challenging because ethanol strongly shapes beer flavor and mouthfeel. Biological production methods, which restrict alcohol formation during fermentation, offer advantages over physical methods, which remove alcohol after fermentation. However, they are limited by the performance and diversity of available yeasts. Recent advances in bioprospecting, breeding, hybridization, adaptive evolution, and genetic engineering reveal how different yeast lineages and trait-development strategies can balance reduced ethanol production with desirable aromatic complexity. Yeasts with naturally limited sugar utilization and favorable aroma‑forming capacities are emerging as promising foundations for next‑generation NAB production. Progress in this field will depend on integrating biological strategies with standardized phenotyping, deeper exploration of natural diversity, and emerging computational tools for predicting flavor and guiding targeted strain improvement. Together, these developments outline a path toward rational, data‑driven design of superior yeasts tailored for NAB production.
|
|
Scooped by
mhryu@live.com
May 31, 9:50 AM
|
Conventional methods for monitoring toxic heavy metals typically require sophisticated laboratory instrumentation and leave a critical gap for rapid, on-site detection. Herein we present Transcription-factor-Occluded Nick Extension or TONE, a rapid, isothermal biosensing platform for heavy metal detection that exploits allosteric transcription factor (aTF) regulation to gate DNA strand-displacement amplification. In this approach, operator sequences modified with deoxyinosine (dI) substitutions are employed. When bound by an aTF, the dI sites are shielded from cleavage by Endonuclease V. Upon exposure to target heavy metals, the aTF dissociates, permitting the enzyme to nick the DNA and trigger a strand-displacement reaction. The amplified DNA is then detected via an instrument-free lateral flow assay, delivering a visual readout within 25 minutes at room temperature. We demonstrate the utility of TONE using the TetR aTF and further adapt the assay for copper and lead detection through the transcription factors CsoR and CadC, respectively, achieving detection limits as low as 40 nM and 80 nM, respectively. TONE offers a sensitive, low-cost, and field-deployable solution for environmental monitoring and other applications requiring rapid heavy metal analysis.
|
|
Scooped by
mhryu@live.com
May 31, 9:34 AM
|
This study presents a mathematical framework for investigating the dynamics of coexistence and competition among heterotrophic microbes across different time scales. Focusing on metabolic interactions, we examine how three strategies: public metabolizing, private metabolizing, and cheating, shape population behavior. The framework integrates generalized Lotka-Volterra dynamics with evolutionary game theory to capture the effects of resource exchange, particularly glucose made available by public metabolizers and sucrose as a shared substrate driving population growth. Game-theoretic payoffs encode ecological costs and benefits, enabling analysis of frequency-dependent interactions among strategies. To capture evolutionary realism, we implement laboratory-inspired simulations in which strategies can switch between generations, mimicking mutation or phenotypic plasticity in microbial populations. These eco-evolutionary dynamics reveal conditions under which all three strategies coexist at interior equilibria and show how variation in growth advantages and, illustratively, phenotype-switching perturbations produce evolutionary shifts. Numerical analysis identifies ecological thresholds and fitness asymmetries that determine system robustness, long-term coexistence, and the persistence of a synthetic, cross-kingdom system linked by nutrient exchange. Together, these insights provide general principles for microbial coexistence and offer design guidelines for ecosystem engineering, biotechnological applications, and the construction of stable synthetic communities under ecological and evolutionary constraints.
|
|
Scooped by
mhryu@live.com
May 31, 1:06 AM
|
The RNA polymerase that transcribes photosynthetic genes in the plant chloroplast is the largest known transcription enzyme across all domains of life, comprising 21 subunits of bacterial and eukaryotic origin. Recent structural analyses revealed that the core polymerase, inherited from the cyanobacterial ancestor of the chloroplast, is encased by subunits unlike known bacterial transcription proteins. This insight into the composite nature of the complex provides clues about how the polymerase interacts with transcription regulatory elements of both bacterial and eukaryotic origin. Here, we summarize insight from recent structural and biochemical data on chloroplast and cyanobacterial transcription complexes. We analyze the available evidence on the mechanisms of chloroplast Sigma factors, NusG, mTERF proteins, kinases and alarmones, considering prevailing models of transcription control in bacteria and mitochondria to produce new hypotheses on the molecular basis of chloroplast transcription control to be characterized in the future.
|
|
Scooped by
mhryu@live.com
May 31, 12:59 AM
|
Prokaryotes harbor a diverse spectrum of extrachromosomal elements (ECEs), which are intracellular replicons maintained independently of the primary chromosome. Historically, the ECE research field has focused on relatively small ECEs, such as plasmids. However, the advent of long-read sequencing has revealed that prokaryotes also harbor various types of giant ECEs, spanning hundreds of kilobases to over 1 Mb, that were not hitherto recognized. In this review, we describe how long-read sequencing has enabled the discovery of giant ECEs and compare the genetic architectures and functional repertoires of several recently characterized examples. The functions of most genes in these ECEs remain uncharacterized, and current computational tools frequently misclassify or overlook them. We further discuss how the discovery of these giant ECEs challenges existing classification frameworks that attempt to distinguish megaplasmids, chromids, and chromosomes. Together, these findings highlight giant ECEs as a largely unexplored layer of microbial genetics, whose characterization will have broad implications for our understanding of microbial adaptation and horizontal gene transfer.
|
|
Scooped by
mhryu@live.com
May 31, 12:37 AM
|
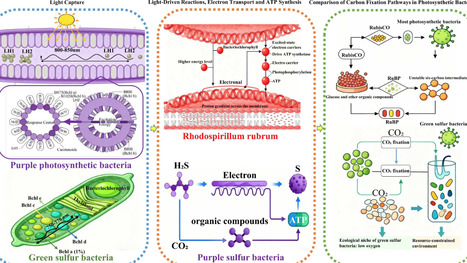
Photosynthetic bacteria, a diverse group of microorganisms endowed with distinctive photosynthetic mechanisms, have attracted increasing attention on account of their substantial ecological functions and extensive application prospects. This review comprehensively summarizes the classification, physiological and biochemical characteristics of photosynthetic bacteria, and explores their pivotal functions in carbon, nitrogen, and sulfur cycling. Moreover, the potential applications of photosynthetic bacteria in biotechnology, including wastewater treatment, agriculture, aquaculture, and bioenergy production, are systematically presented. The latest research methods, current research trends, and future development directions are also analyzed in detail. Despite their considerable potential, challenges like high-cost large-scale cultivation and strain instability still exist. A more comprehensive understanding of photosynthetic bacteria will contribute to their more effective utilization in addressing environmental and energy problems.
|