 Your new post is loading...

|
Scooped by
mhryu@live.com
Today, 11:56 AM
|
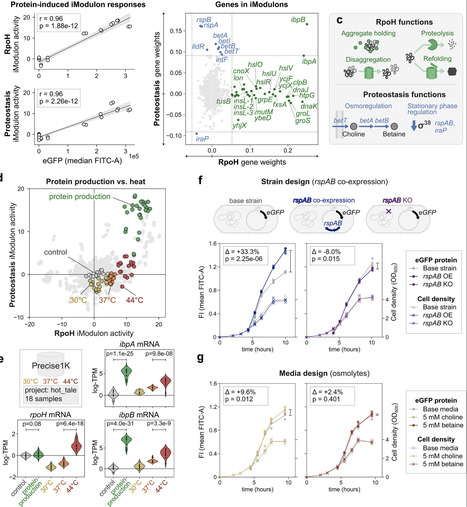
Predictable expression of heterologous genes remains a key challenge in biotechnology, largely due to cellular stresses imposed on the production host. Here, we systematically dissect stress responses in E. coli MG1655 expressing diverse proteins under varying promoters and translation efficiencies. Using independent component analysis on new and existing large transcriptomic datasets, we identify distinct responses to transcriptional and translational stresses: excessive heterologous mRNA triggers a cold shock response that controls mRNA stability (cspA–I, deaD), while protein production activates a heat shock response involving proteolysis and chaperone functions. We further identify a broad adaptation response, consistently co-activated with the heat shock response during protein production, that provides stationary phase regulation (rspAB) and osmoregulation (betABIT). Targeting these latter functions through strain and media modifications significantly increases eGFP production. Other host stress responses depend on the protein being expressed; e.g. we find production of cysteine-rich proteins to uniquely activate functions regulating iron- and redox homeostasis and oxidative stress responses. This work demonstrates a holistic, systems-level view of cellular stresses to heterologous gene expression by considering transcriptional, translational, and product-specific contributions, paving the way toward predictable and optimized expression strategies.
|
|
Scooped by
mhryu@live.com
Today, 11:45 AM
|
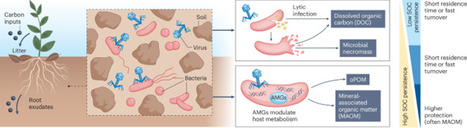
Growing evidence indicates that soil viruses influence carbon cycling by modulating microbial metabolism and infection dynamics through auxiliary metabolic genes. Beyond host cell lysis, viral processes might regulate carbon flow and the formation of stabilized carbon pools. Integrating viral ecology into soil carbon models and restoration strategies is essential.
|
|
Scooped by
mhryu@live.com
Today, 10:48 AM
|
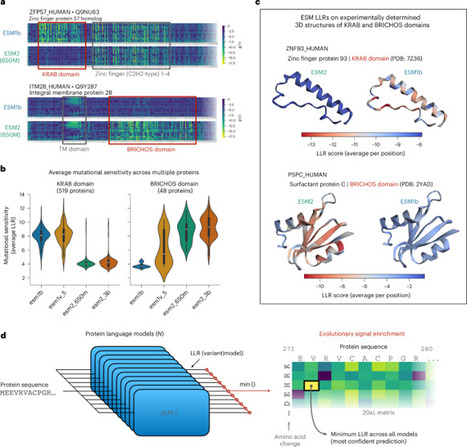
Protein language models (PLMs) have recently emerged as a promising approach for next-generation variant-effect prediction (VEP). Most high-performing VEP methods currently utilize PLMs combined with additional information, such as homology, protein structure and population genetics data to improve prediction accuracy. This performance gain, however, comes with added complexity or limited applicability compared to pure PLMs trained only on raw, unaligned sequences, such as evolutionary scale modeling (ESM). Here we challenge the prevailing view that sequence-only PLMs are intrinsically limited and present an efficient co-distillation approach to adapt them for high-accuracy VEP without requiring additional information beyond evolutionary signals captured during pretraining. We allow individual PLMs to self-improve by distilling the most confident predictions from multiple models of the same family and demonstrate that co-distillation of ESM models suffices to achieve state-of-the-art performance across multiple VEP benchmarks. We further show that this performance increase enables accurate quantification of the severity of variant effects on continuous clinical phenotypes in biobank data. A co-distillation framework is used to iteratively adapt sequence-only protein language models for high-accuracy variant effect prediction, without the need for additional structural or genetic data. Individual protein language models therefore self-improve by distilling the most confident predictions from multiple models, achieving state-of-the-art performance across multiple variant effect prediction benchmarks.
|
|
Scooped by
mhryu@live.com
Today, 10:37 AM
|
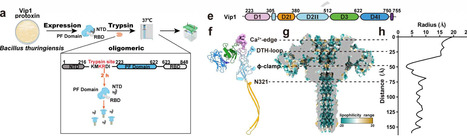
Insecticidal toxins from Bacillus thuringiensis (Bt) have been extensively and successfully used in genetically engineered crops for decades but continue to face challenges from the adaptive resistance in the insect population. The Bt binary toxin Vip1Ad1(Vip1) and Vip2Ag1(Vip2), a promising next-generation candidate gene combination for transgenic crops, have demonstrated high efficacy against the destructive coleopteran pest white grubs, however, their mode of action remains largely elusive. In this study, we report cryo-EM structures of the heptameric Vip1-pore and Vip2-bound Vip1-pore complex, capturing a series of putative assembly-related intermediates that suggest a binary toxin assembly and translocation pathway. Together with structure-guided mutagenesis, these data provide insights into a sequential assembly of binary complex and a sequence-independent translocation mechanism. Proof-of-principle experiments showed successful delivery of a desired protein cargo into host cells based on the mini-Vip2-Vip1 pore system, paving the way for developing much needed extracellular pesticidal protein delivery platforms. These findings not only clarify the assembly and translocation mechanism of the binary insecticidal toxin pair but also offer an excellent alternative model to investigate human-pathogenic pore-forming toxins because of its similarity and biosafety. This study reveals how the Vip1-Vip2 binary toxin assembles and enables protein translocation into target cells, providing structural insights into binary toxin action and suggesting strategies for developing next-generation bioinsecticides.
|
|
Scooped by
mhryu@live.com
March 29, 5:51 PM
|
Motility often plays a decisive role in the survival of species. Five systems of motility have been studied in depth: those propelled by bacterial flagella, eukaryotic actin polymerization and the eukaryotic motor proteins myosin, kinesin and dynein. However, many organisms exhibit surprisingly diverse motilities, and advances in genomics, molecular biology and imaging have showed that those motilities have inherently independent mechanisms. This makes defining the breadth of motility nontrivial, because novel motilities may be driven by unknown mechanisms. Here, we classify the known motilities based on the unique classes of movement-producing protein architectures. Based on this criterion, the current total of independent motility systems stands at 18 types. In this perspective, we discuss these modes of motility relative to the latest phylogenetic Tree of Life and propose a history of motility. During the ~4 billion years since the emergence of life, motility arose in Bacteria with flagella and pili, and in Archaea with archaella. Newer modes of motility became possible in Eukarya with changes to the cell envelope. Presence or absence of a peptidoglycan layer, the acquisition of robust membrane dynamics, the enlargement of cells and environmental opportunities likely provided the context for the (co)evolution of novel types of motility.
|
|
Scooped by
mhryu@live.com
March 29, 5:44 PM
|
Precise control of gene expression in a cell-state-specific manner is essential for effective therapeutic interventions in complex and dynamic disease microenvironments. Traditional targeting strategies that rely on surface markers or cell type-specific promoters often assume static cellular identities, limiting effectiveness in context such as cancer and inflammation, where cell states are highly heterogeneous and dynamic. RNA sensors, such as RADAR (RNA sensing using Adenosine Deaminases Acting on RNA), provide a modular, programmable, and nonintegrating platform for classifying cell states. However, it is also characterized by low sensitivity and dynamic range, which limits its applications in detecting low-abundance transcripts. In this work, we integrate RADAR sensors with a signal amplification circuit to enhance sensitivity and dynamic range. We demonstrate that this combined RADAR-amplifier platform enables real-time monitoring of subtle changes in the abundance of endogenous transcripts under physiological conditions. Our results demonstrate the utility of this platform for fundamental biological studies and the development of precision therapeutic strategies.
|
|
Scooped by
mhryu@live.com
March 29, 5:32 PM
|
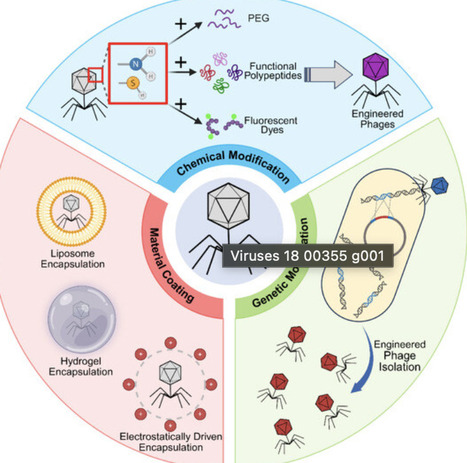
The escalating crisis of antimicrobial resistance (AMR) and the stagnating antibiotic pipeline have renewed interest in bacteriophage therapy. While natural phages offer specificity and low toxicity, their narrow host range, bacterial resistance, and safety concerns limit clinical use. To overcome these hurdles, phages are being engineered using biotechnology. This review outlines the history of phage therapy and systematically summarizes advances in engineered phage preparation, including genetic modification, chemical conjugation, and physical encapsulation. We highlight the application of engineered phages against multidrug-resistant infections, gastrointestinal diseases through gut microbiome modulation, and as targeted delivery vehicles or immune adjuvants in cancer therapy. While significant advances have been made, several critical challenges remain, particularly in regulatory approval, large-scale manufacturing, and ensuring long-term safety. We conclude that engineered phages, as customizable and precise biological tools, are poised to advance precision phage medicine, offering a transformative solution to AMR and fostering convergence across synthetic biology, medicine, and environmental science.
|
|
Scooped by
mhryu@live.com
March 29, 3:53 PM
|
T7 RNA polymerase is a foundational enzyme for biotechnology, but its utility for many potential applications is limited by low thermal stability of 43-44°C. While stabilized variants exist, the most stable commercial version has a proprietary sequence. In this work we developed a highly stable T7 RNAP using structure-based computational design. We combined mutations from previous stabilized variants (M5, M8, V7abcd) with new mutations identified by PROSS. These mutations were filtered using data-driven heuristics to preserve function. Our final design, T7T+, contains 30 point mutations from the original T7 RNAP and demonstrates a functional stability (T50) of 54.9°C in a thermal challenge assay, which is 2.4°C higher than the most stable, published open-source variant to date. Circular dichroism spectroscopy showed an apparent melting temperature of 53.8°C. T7T+ retains 59% of wild-type activity at 37°C. 16 of the 18 tested protein designs had higher stability against thermal challenge compared with the genetic background, attesting to the high success rates of existing non deep learning computational methods for the design of stable, functional proteins. A plasmid encoding T7T+ has been deposited in AddGene and is freely available for non-commercial use.
|
|
Scooped by
mhryu@live.com
March 29, 3:43 PM
|
Diatoms exhibit high competitive capacity in nitrogen assimilation, but the underlying mechanisms remain unclear. Here, we identify a non-ribosomal peptide synthase-like gene (PtNRPS1) with an atypical domain structure (A-T-R1-R2) in the marine diatom Phaeodactylum tricornutum, crucial for short-term nitrogen assimilation. In vitro enzyme assays show PtNRPS1 catalyzes conversion of L-tryptophan to tryptophanol, a tryptophan-derived indole compound that promotes diatom growth at concentrations far lower than indole-3-acetic acid. Transcriptomic, metabolomic analyses, and stable-isotope analyses indicate tryptophanol enhances short-term nitrogen assimilation. CRISPR-Cas9 knockout of PtNRPS1 abolishes tryptophanol biosynthesis and reduces nitrogen-assimilation enzymes activities, which are restored by exogenous tryptophanol. PtNRPS1 overexpression results in delayed but sustained enzyme elevation. Global distribution of PtNRPS1 homologues in stramenopiles positively correlates with nitrogen-assimilation gene abundance. Our findings suggest tryptophanol, synthesized by a diatom NRPS, accelerates nitrogen assimilation, providing a competitive edge in oceanic nitrogen acquisition. . Here the authors show that marine diatoms produce tryptophanol, a molecule that at extremely low concentrations rapidly activates nitrogen assimilation genes and enzymes. This facilitates acquisition of nitrogen, a vital ocean nutrient.
|
|
Scooped by
mhryu@live.com
March 29, 3:32 PM
|
In all kingdoms of life, the regulation of membrane-bound enzyme function via oligomerization is a fundamental aspect of cell physiology. Often, the mechanistic role of oligomerization is unclear, due to a lack of structure-function comparisons between constituent forms of the enzyme. Here, we elucidate the structural underpinnings of enzyme regulation and oligomerization in the quinol-dependent nitric oxide reductase (qNOR) from Neisseria meningitidis, by high-resolution structural analyses of the less active monomeric form (2.25 Å) and the highly active dimeric form (1.89 Å). The comparison revealed that broad helical flexibility near the dimer interface of the monomer causes a conformational change in a critical amino acid near the active site, located apart from the dimer interface. We demonstrate that the crosstalk between the dimer interface and catalytic site in qNOR allows enhanced activation of the enzyme via dimerization. Given Neisseria meningitidis’ dependence on qNOR to detoxify the host’s immune response of nitric oxide, our results pave a way for new strategies to combat bacterial infections, via the inactivation of qNOR by monomerization. More broadly, this provides new insights into the role of membrane protein oligomerization and its influence on regulating activity. Dimer-monomer structural and functional comparison of quinol-dependent nitric oxide reductase from Neisseria meningitidis reveals insights into the interlinking role of dimer stability and high catalytic activity.
|
|
Scooped by
mhryu@live.com
March 29, 3:14 PM
|
CRISPR-based epigenome editing represents a programmable strategy to precisely modulate gene expression, holding immense promise for therapeutic applications. However, the large size of the dCas proteins substantially impedes the delivery via adeno-associated virus (AAV) vectors. Here, through iterative bioinformatics analysis, structure-guided predictions, and functional assays, we identified and characterized a novel miniature subtype V-M CRISPR-Cas12m from Pelomicrobium methylotrophicum. PmCas12m exhibited flexible 5'-YTN-3' PAM-dependent recognition and robust double-stranded DNA binding properties, while lacking DNA cleavage activity, thus positioning it as an ideal tool for epigenome editing. Cryogenic electron microscopy (cryo-EM) structures of PmCas12m unveiled its unique molecular mechanism of DNA binding facilitating interference. Guided by these structural insights, we employed deep mutational scanning (DMS) and protein engineering to develop xCas12m, a hypercompact variant with highly potent and specific epigenome editing capabilities in human cells. We further constructed the xCas12m-CRISPRoff platform in a single AAV vector, which achieved durable epigenetic silencing and effective inhibition of hepatitis B virus (HBV) infection in a mouse model. Collectively, these findings establish xCas12m as a versatile epigenome editing platform with transformative potential for treating diseases, paving the way for clinical translation of epigenetic therapies.
|
|
Scooped by
mhryu@live.com
March 29, 2:45 PM
|
Cabbage black rot, caused by Xanthomonas campestris pv campestris (Xcc), is a severe disease worldwide. The predominant method for controlling this disease relies on agrichemicals, which increase antibiotic resistance. This study showed that the secondary metabolites of B. cereus BR3 elicited induced systemic resistance in cabbage and impaired the type III secretion system (T3SS) of Xcc. Furthermore, the extract of strain BR3 significantly enhanced the activities of plant-defense-related enzymes, suppressed the hypersensitive response (HR) induced by Xcc in tobacco, and attenuated the pathogenicity of Xcc on cabbage. Additionally, several genes of Xcc T3SS were repressed by the BR3 extract. Activity-guided purification identified 3-methylcinnamic acid as an effective inhibitor against the T3SS of Xcc. Overall, this study suggests that 3-methylcinnamic acid produced by strain BR3 disrupts a critical virulence mechanism in Xcc, providing a promising alternative for controlling the black rot disease in cabbage.
|
|
Scooped by
mhryu@live.com
March 29, 2:36 PM
|
The first and arguably most critical level of gene expression is transcription of the genetic information from DNA into RNA. Within this process, transcription initiation stands out as the key step that influences all downstream events. Central to initiation are promoters, DNA sequences that interact with the key enzyme of transcription, RNA polymerase. A single transcription unit may be controlled by one or multiple promoters, a strategy found across all domains of life. This review highlights how combinatorial promoter arrangements control gene expression in microorganisms, with brief comparisons to other organisms. We also explore how these promoter architectures function as sensors for stress-related molecules, such as antibiotics. We examine how these insights can be applied to predict previously unidentified mechanisms of antibiotic resistance. Finally, the use of dual-promoters in synthetic biology is outlined and discussed.
|
|
|
Scooped by
mhryu@live.com
Today, 11:53 AM
|
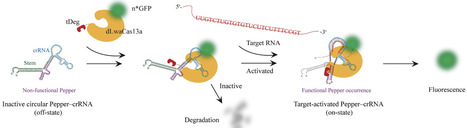
RNA molecules display remarkable heterogeneity in structure, dynamics, and function, yet methods for their precise visualization in living cells remain limited. While CRISPR-based RNA imaging holds great potential, existing systems often suffer from high background fluorescence due to constitutive signal emission or non-specific binding. To overcome these challenges, we developed CtDeg (CRISPR–dCas13–tDeg), a modular RNA imaging platform that links fluorescence activation directly to target RNA recognition while leveraging degron-mediated degradation to suppress background signals. By engineering the crRNA scaffold to embed the Pepper RNA motif, CtDeg ensures that fluorescence is present only upon binding to the native RNA target. We systematically optimized C-terminal tDeg variants to maximize the signal-to-noise ratio and demonstrated that CtDeg achieves substantially lower background and higher specificity than conventional fluorescent protein–CRISPR-based RNA imaging approaches. Using CtDeg, we captured real-time paraspeckle assembly dynamics and visualized early-stage SARS-CoV-2 genomic RNA transport. Remarkably, CtDeg provided the first direct imaging evidence of virus-induced NEAT1_2 lncRNA accumulation, revealing a host-virus regulatory interaction. Beyond these applications, CtDeg is compatible with multiple Cas13 orthologs and fluorescent proteins, establishing a versatile, target-induced platform for probing RNA localization, dynamics, and function in living cells, with broad applications in synthetic biology and RNA biology.
|
|
Scooped by
mhryu@live.com
Today, 10:57 AM
|
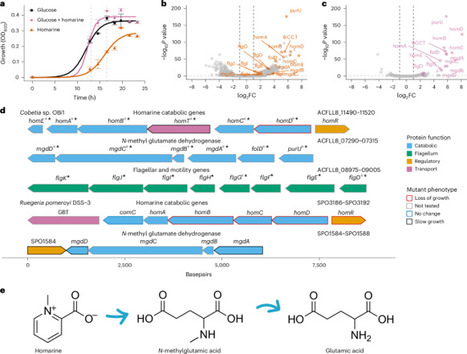
Homarine (N-methylpicolinic acid) is a ubiquitous marine metabolite produced by phytoplankton and noted for its infochemical properties among marine animals, yet its microbial degradation pathways are uncharacterized. Here we identify a conserved operon (homABCDER) that mediates homarine catabolism in bacteria using comparative transcriptomics, mutagenesis and targeted knockouts. Phylogenetic and genomic analyses show that this operon is distributed across abundant bacterial clades, including coastal copiotrophs (for example, Rhodobacterales) and open-ocean oligotrophs (for example, SAR11, SAR116), and in genomes from non-marine environments. High-resolution mass spectrometry revealed N-methylglutamic acid and glutamic acid as key metabolic products of homarine in both model and natural systems, with N-methyl-glutamate dehydrogenase catalysing their conversion. Metatranscriptomics showed responsive and in situ expression of hom genes aligned with homarine availability. These findings uncover the genetic and metabolic basis of homarine degradation, establish its ecological relevance and highlight homarine as a versatile growth substrate that feeds into central metabolism via glutamic acid in diverse marine bacteria. Homarine is a ubiquitous, phytoplankton-derived metabolite that is broken down by widely distributed and diverse marine bacteria containing a conserved homABCDER operon.
|
|
Scooped by
mhryu@live.com
Today, 10:38 AM
|
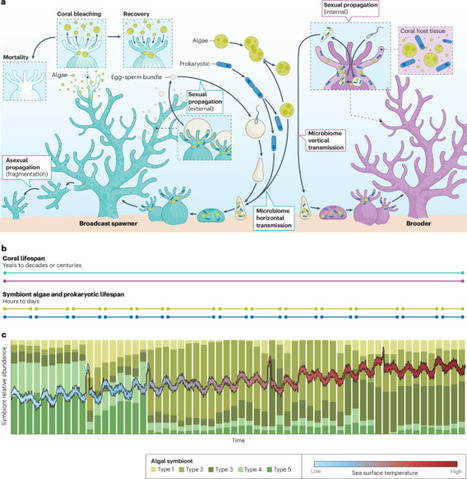
Coral reefs are increasingly threatened by marine heatwaves, prompting the need for proactive interventions that enhance coral thermal tolerance. Assisted evolution, which aims to accelerate natural adaptation rates, has emerged as a promising approach. However, programmes of assisted evolution must outpace the escalating frequency and intensity of marine heatwaves. Here, we present a Roadmap for accelerating progress towards using assisted evolution to enhance coral thermal tolerance. We highlight advances in coral biology across cellular, organismal, and ecological scales that support the feasibility of assisted evolution in coral populations. We compare current experimental gains in thermal tolerance via assisted evolution with projected temperatures, finding that these are unlikely to keep pace with predicted climate change. We identify key knowledge gaps that hinder timely development of assisted evolution and propose a comprehensive research agenda to address these gaps. This agenda will be catalysed by large-scale, multi-institutional field hubs increasing experimental scope and statistical power, support for long-term research at these hubs, spanning coral generations, and development and application of methodologies that safeguard broodstock and experimental corals from disturbances. By implementing these proposals, scientists can realize the potential of assisted evolution and help to safeguard a future for coral reefs. Ongoing and projected climate changes are bringing increased marine heatwave frequency and intensity, threatening the health and survival of coral reefs. This Roadmap outlines the potential for assisted evolution methods to increase thermal tolerance in corals and describes ways to accelerate research and development for enhancing coral adaptation rates.
|
|
Scooped by
mhryu@live.com
Today, 10:20 AM
|
This Genome Watch article explores how taking into account known phylogenetic relationships can improve computational efficiency for genomics, enabling improved genome data compression and faster sequence search as datasets continue to expand. genome compression, zip
|
|
Scooped by
mhryu@live.com
March 29, 5:48 PM
|
Actinobacteria represent a prolific source of bioactive natural products. However, the complex transcriptional regulatory networks in these bacteria, particularly the interplay between transcription factors (TFs) and their regulatory ligands (TF-RLs), remain poorly characterized and lack dedicated resources. In this context, we introduce the Actinobacteria Transcription Factor Database (Actinobacteria TFDB), a comprehensive repository that systematically integrates TF-centric data across 25 representative species. The current version encompasses 629 TFs, classified into 69 families, documents 11,776 TF-target relationships and 28 TF posttranslational modification sites. Uniquely, it features a dedicated collection of 54 experimentally validated TF-RL interactions. Beyond providing standardized annotations, sequence and structural features, and regulatory networks, Actinobacteria TFDB incorporates a specialized TF-RL module that enables interactive exploration and visualization of allosteric regulatory mechanisms. By consolidating multi-dimensional TF data from diverse sources, this resource empowers systems-level analyses and facilitates the rational design of regulatory strategies to activate silent biosynthetic gene clusters and optimize metabolite production. The database is publicly available at http://mingleadgene.com:9315/#/home.
|
|
Scooped by
mhryu@live.com
March 29, 5:38 PM
|
In vivo multiple-enzyme cascades have attracted considerable interest for their ability to provide a native microenvironment that supports enzymatic activity and membrane protein function. This review outlined four pivotal strategies for their optimization, increasingly empowered by Artificial Intelligence (AI): (1) enhancing enzyme performance by enzyme discovery and engineering; (2) precisely modulating enzyme expression via rationally designed genetic regulatory elements; (3) implementing spatial and stoichiometric control using protein, nucleic acid, or synthetic scaffolds and compartments; and (4) employing multimodule systems including multiple cell modules and hybrid in vivo/in vitro cascades. Advances in AI accelerate these strategies, enabling novel approaches such as de novo protein design, directed evolution, and the computational design of genetic parts and supramolecular scaffolds. The integrated implementation of these methods substantially increased target compound titers. This lays a strong foundation for industrial implementation. However, several key challenges remain to be addressed.
|
|
Scooped by
mhryu@live.com
March 29, 5:24 PM
|
Exosomes, despite their promise as drug carriers for crossing biological barriers, remain underexplored for noninvasive posterior ocular delivery. Here, we demonstrate that semen-derived exosomes (SEVs) penetrate ocular barriers effectively, owing to their epidermal growth factor expression, which mediates reversible tight-junction disruption. SEVs reach the posterior segment via dual corneal and conjunctival routes. Using this, we engineered FA-SEVs@CMG eye drops, where SEVs are modified with folic acid (FA) and loaded with a nanozyme system (CMG) composed of carbon dots, manganese dioxide, and glucose oxidase. This eye drop leverages SEVs’ excellent penetration ability and FA’s targeting effect to enhance drug delivery to retinoblastoma (RB) cells. Internalized CMG induces intense oxidative stress, disrupts the autophagy-apoptosis balance, and triggers RB cell self-destruction. In vivo, FA-SEVs@CMG effectively inhibits RB growth while preserving retinal function. This work establishes the first SEV-based platform for noninvasive posterior segment delivery, offering a transformative strategy for treating posterior ocular diseases.
|
|
Scooped by
mhryu@live.com
March 29, 3:45 PM
|
Formic acid (FA), a key one-carbon liquid compound derived directly from CO2, can serve as a dual-purpose substrate in microbial metabolism, supplying both carbon and energy. Its potential for green biomanufacturing is immense, yet its inherent toxicity and poor metabolizability to most microbes pose a major hurdle in developing efficient microbial cell factories for value-added chemical production. Building on our prior discovery of Vibrio natriegens as a naturally proficient formic acid utilizer, we demonstrate here that formate supplementation as an auxiliary substrate can dramatically boost pyruvate production of V. natriegens from sodium gluconate, achieving a 1.9-fold increase in titer. Transcriptomic analysis revealed that formate presence induces global changes in gene expression. By subsequently downregulating the pyruvate consumption pathway, we engineered a strain that, when co-fed with formate and sodium gluconate, achieved a 49.0% improvement in pyruvate synthesis. Isotopic tracer analysis confirmed substantial formate assimilation, with approximately 9.43% incorporated into biomass. In a fed-batch fermentation, the engineered V. natriegens strain consumed 82.8 g/L sodium gluconate and 37.4 g/L formate (HCOONa·2H2O) within 51 h, producing 56.4 g/L pyruvate at a rate of 1.1 g/L/h. This work elucidates the stimulatory role of formate in the pyruvate biosynthesis of V. natriegens and establishes a novel strategy for leveraging this feedstock in microbial production.
|
|
Scooped by
mhryu@live.com
March 29, 3:38 PM
|
Bacteriophages are key drivers of microbial ecology, co-existing and co-evolving with bacteria across diverse environments. Limitations in culturing, alongside advances in sequencing and bioinformatics, have driven the use of metagenomics to explore viral diversity. Viral-specific analysis of >3000 food metagenomes from cFMD produced the FVGC, comprising ~3400 metagenome-assembled viruses, most of which belong to novel Caudoviricetes lineages (n = 91), with only ~15% represented in IMG/VR v4. Together, these findings reveal extensive uncharacterized viral diversity in food systems. Beyond serving as a reference, the FVGC facilitates detailed investigation of virus–host interactions. Viral sequences were pervasive across microbial genomes, with several bacterial families exhibiting near-universal associations with viral elements. Bacterial antiviral defence systems were abundant and taxonomically diverse, dominated by restriction–modification systems, while CRISPR–Cas systems showed pronounced lineage-specific distributions; in contrast, viral anti-defence genes were detected at low frequency (<10% of MAVs). Host prediction linked MAVs to clinically relevant taxa, including expanded ESKAPE pathogens such as Klebsiella pneumoniae, Acinetobacter baumannii, Staphylococcus aureus, and Enterobacter spp., highlighting the ecological connectivity between food-associated viruses and clinically important bacteria. Antimicrobial resistance signals were scarce, suggesting minimal phage-mediated AMR dissemination in food environments. This new publicly available viral database represents a valuable resource for further exploration of viral diversity.
|
|
Scooped by
mhryu@live.com
March 29, 3:21 PM
|
Microplastics (MPs) in wastewater treatment plants (WWTPs) represent novel ecological niches rather than passive contaminants. They rapidly acquire distinct microbial biofilms, forming the “Plastisphere”. This Perspective examines the underexplored role of quorum sensing (QS) within these complex synthetic microenvironments. We highlight how the intrinsic properties of MPs, such as surface hydrophobicity, chemical leachates, and aging states, create altered signaling landscapes. These distinct environments may decouple QS responses from classical population-density thresholds. Consequently, these dynamic interactions potentially enhance cooperative microbial metabolism and may indirectly facilitate the horizontal gene transfer of antibiotic resistance genes by fostering dense cellular proximity. Despite substantial methodological challenges in detecting active signaling in situ, MPs must be reconceptualized as quorum-modulating microscopes capable of reprogramming microbial communication. Advancing this field requires integrating high-resolution spatial imaging and functional genomics under operationally relevant conditions to bridge theoretical insights with empirical validation, ultimately informing future treatment designs and environmental risk assessments. Microplastics intensify quorum-sensing-mediated gene exchange in wastewater treatment plants, enabling their reconceptualization as quorum-modulating microscopes that reprogram microbial communication and function, based on their interplay with biofilms and quorum sensing within wastewater systems
|
|
Scooped by
mhryu@live.com
March 29, 3:10 PM
|
Direct Coupling Analysis has been instrumental over the past decade in leveraging evolutionary information and advancing our understanding of biomolecular structure and function. Here, we introduce sparse extensions of this method that explicitly incorporate structural information. StructureDCA focuses on physically relevant interactions by selectively retaining couplings between residues in spatial contact, and StructureDCA[RSA] additionally incorporates per-residue relative solvent accessibility. These models outperform state-of-the-art approaches in describing mutational landscapes, as they more effectively integrate structural context. Moreover, their sparse formulation enables orders-of-magnitude improvements in computational efficiency while preserving interpretability, providing a powerful framework for gaining mechanistic insights into mutation effects and advancing protein design. The StructureDCA models are available as a user-friendly Python package via the PyPI repository. The source code is freely accessible at https://github.com/3BioCompBio/StructureDCA, which also includes a Colab Notebook interface.
|
|
Scooped by
mhryu@live.com
March 29, 2:43 PM
|
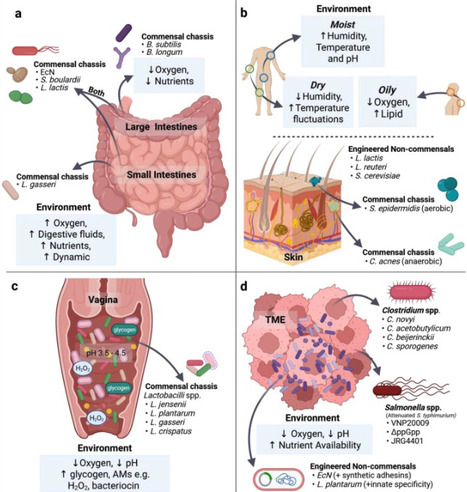
The human microbiota comprises a vast and diverse array of microorganisms that play critical roles in maintaining health and modulating diseases. Engineered live biotherapeutic products (eLBPs) harness genetically modified microbes to perform defined therapeutic functions within the host. A central challenge in developing effective eLBPs is the rational selection of an appropriate microbial chassis, which requires consideration of safety, genetic tractability, functional performance and the target host niche. As different body sites present distinct physiological and biochemical conditions that influence microbial survival, colonisation and activity, choosing a chassis well-adapted to the target niche is essential for therapeutic durability and sustained efficacy. This review summarises microbial chassis that have been widely employed for the gut, skin, vagina and tumour microenvironment, highlighting how their characteristics enable effective function within these niches. Strategies to improve eLBP colonisation within these niches are discussed, and emerging microbial candidates that hold promise as future eLBPs are also identified.
|


























mining, Utilizing MmCas12m (WP_061006603.1) as an initial query, we performed a PSI-BLAST search against the NCBI non-redundant database and then constructed multiple HMM profiles of the corresponding representative protein domain architecture based on the sequence alignments. we employed AlphaFold 3 to predict the 3D structures of the CRISPR-Cas12m candidates and compared these predicted structural architectures with that of MmCas12m using the DALI web server