Your new post is loading...

|
Scooped by
mhryu@live.com
March 26, 11:47 PM
|
The canonical amino acid alphabet has remained remarkably stable since life’s early stages, yet the factors that shaped its emergence remain debated. Early views emphasized prebiotic availability and the expansion of metabolic pathways, but recent advances (particularly from protein biophysics and deep phylogenetics) have brought new perspectives to this question. Together, these views agree that the canonical alphabet emerged from a chemically restricted repertoire and was gradually tightened by metabolic innovation and selection for foldable, functional proteins. However, these approaches can yield inconsistent trajectories, underscoring how much remains unresolved. Here, we compare insights from four lines of evidence, highlight their limitations, and argue that our chronology of amino acid recruitment should be based on where the approaches converge.
|
|
Scooped by
mhryu@live.com
March 26, 11:09 PM
|
Bacterial surface polysaccharides are versatile structures that provide specificity to the behavior and interactions of a given bacterial strain. One surface polysaccharide displayed on the organism Bacteroides fragilis, Capsular Polysaccharide A, has been implicated as a potential therapeutic for autoimmune disorders. This polymer is composed of repeating units of the tetrasaccharide 2-acetamido-4-amino-2,4,6-trideoxygalactopyranose (AATGal), 4,6-O-pyruvate-galactopyranose (PyrGal), N-acetylgalactosamine (GalNAc), and galactofuranose (Galf). While this and other bacterial surface polysaccharides are attractive to study and apply to biomedicine, it can be difficult to acquire quick, inexpensive access to these pure materials. In this work, we developed a recombinant expression system in E. coli for the stepwise production of the CPSA polymer. A series of sequential plasmids were prepared, each incorporating successive genes required for CPSA biosynthesis. Using these iterative plasmids, we were able to observe production of the CPSA repeating unit and precursors by liquid chromatography mass spectrometry (LC-MS) analysis of cell lysates. We found that it was critical to include the CPSA polymerase but not the flippase, indicating that a native E. coli flippase could support polymer production. We also provide evidence that the CPSA polymer produced by E. coli can be ligated to LPS by the E. coli WaaL ligase, and deletion of this gene led to the formation of a water-soluble polymer. Overall, this work describes the first recombinant system for CPSA production and outlines a key strategy for the production of complex glycopolymers.
|
|
Scooped by
mhryu@live.com
March 26, 9:09 PM
|
The assembly of newly synthesized proteins into functionally active oligomers has long been regarded as a posttranslational process driven by random collision of subunits. However, growing evidence indicates that, for many proteins, assembly occurs cotranslationally, tightly coupling synthesis, folding, and subunit assembly. This fundamentally different mechanism enables the spatial and temporal coordination of assembly, promotes the hierarchical formation of multisubunit assemblies, enhances the stability of involved subunits, enlarges the space of feasible protein structures including complexes with intertwined subunits, and has profound effects on protein evolution and function. In this review, we describe the molecular mechanisms, cellular requirements, and functional implications of cotranslational assembly and discuss its relevance to human disease, its evolutionary significance, and its transformative potential in synthetic biology and recombinant protein production.
|
|
Scooped by
mhryu@live.com
March 26, 8:46 PM
|
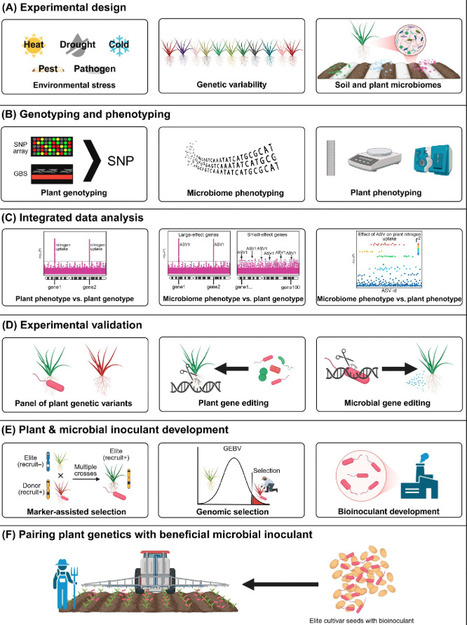
Plants constantly form associations with microorganisms, and some are vital for plant performance, especially under stress conditions. Although some microorganisms have been developed into commercial bioinoculant products, their associations with plants can be transient, and their efficacy is often inconsistent in the field. Here, we propose a framework and key research steps needed for plant breeders and microbiologists to work together to match plant genetics with compatible microbial inoculants. Recent studies have successfully identified plant genetic factors that play a role in the recruitment of beneficial microorganisms, and many associated technologies are ready for implementation towards this goal. This innovative and collaborative approach could provide novel, enduring plant–microbiome associations for environmental sustainability and food security under a changing climate.
|
|
Scooped by
mhryu@live.com
March 26, 8:15 PM
|
Antimicrobial agents play a vital role in human and environmental health, with applications spanning medicine, food preservation, agriculture, and biotechnology. Among them, enzybiotics enzyme-based antimicrobials have emerged as powerful alternatives to conventional antibiotics due to their targeted mechanisms and lower propensity for resistance. Beyond their medical relevance, enzybiotics have emerging applications in food preservation, animal health, and agriculture, thereby broadening their industrial and environmental value. To support the discovery and characterization of these versatile biomolecules, we present the first genome-resolved metagenomic gene and protein targeted enzybiotic catalog focused on enzybiotics, derived from diverse environmental microbiomes. The Microbial Enzybiotic Gene and Protein Catalog (MiGPC), integrates 15 whole-metagenome datasets from oceans, soils, fecal samples, vegetation, and plastic-contaminated environments, capturing a wide ecological spectrum. Enzybiotic sequences were compiled through a hybrid strategy combining public database mining and manual literature curation, yielding over 136,000 enzybiotic sequences, 7654 metagenome-assembled genomes (MAGs), and ~ 100 million unique genes and proteins. MiGPC integrates taxonomic and enzybiotic gene profiles, offering a robust platform for the discovery, annotation, and ecological mapping of antimicrobial enzymes. Functional analyses using KEGG and eggNOG revealed that approximately 62% of the genes remained uncharacterized, highlighting a rich source of potentially novel functions. Glycoside hydrolases and glycosyl transferases were the most prevalent CAZyme families, while the dominant enzybiotic-producing taxa belonged primarily to the Pseudomonadota and Bacillota phyla. Statistical modeling uncovered two major ecological clusters that distinguished polluted from relatively pristine environments. MiGPC enables high-throughput screening of previously unexplored metagenomes, facilitating the identification of novel antimicrobial agents from under characterized ecosystems. Overall, MiGPC represents a landmark resource that will support multi-omics research, microbial ecology, and the development of next-generation biotechnological solutions based on enzybiotics.
|
|
Scooped by
mhryu@live.com
March 26, 7:40 PM
|
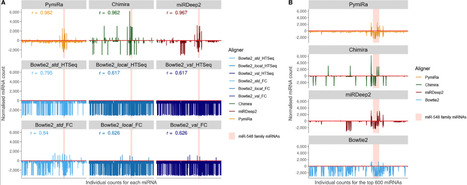
Small non-coding RNAs (sncRNA; < 200 nucleotide length) are of increasing research interest due to their key regulatory roles in a host of fundamental biological processes. For example, microRNAs (miRNAs), a specific class of sncRNAs, regulate gene expression through messenger RNA interactions, and their dysregulation is associated with disease. Classifying sncRNAs is an important bioinformatic task in small RNA-sequencing pipelines. Here we have developed an aligner called PymiRa, written in Python, to identify and quantify miRNAs from FASTA/FASTQ sequencing files. Unlike other approaches, PymiRa utilizes a Burrows-Wheeler algorithm to align an input file against a reference hairpin precursor FASTA file derived from miRBase, the online miRNA registry, permitting up to two mismatches at the 3’ end of a read. Previous tools used either a Burrows-Wheeler genome alignment or dynamic programming alignment to precursors; we demonstrate that combining both approaches yields improved results and efficiency. Importantly, the PymiRa aligner accounts for 3’ post-transcriptional modifications to miRNAs that typically occur. PymiRa is a fast, accurate, and publicly accessible aligner available via GitHub and/or a webserver for sncRNA identification, including miRNAs, enabling accurate counts to be produced as part of a small RNA-sequencing pipeline. PymiRa will undergo relevant revisions over time e.g., with miRBase version updates. The PymiRa aligner will facilitate a deeper biological understanding of the landscape of sncRNA expression in normal physiological conditions and their dysregulation in disease states, including cancer.
|
|
Scooped by
mhryu@live.com
March 26, 7:09 PM
|
The ability to precisely control gene expression is fundamental to studying biological processes. Using site-specific recombinases such as FLP, gene expression can be controlled, albeit with limited spatio-temporal precision. We develop a photocaged FLP recombinase, which can be precisely controlled using light, and we demonstrate its efficacy in Caenorhabditis elegans. We use genetic code expansion to incorporate photocaged amino acids into FLP, replacing critical residues in the active site with their photocaged counterparts. Photocaged FLP displays no detectable background activity, and brief illumination can be used to activate FLP with near 100% efficiency. We show that photocaged FLP can be activated by light between 365 and 435 nm, and that it is not activated by light above 450 nm, making it fully compatible with wavelengths commonly used for imaging and optogenetics. Furthermore, we demonstrate that photocaged FLP can be used to switch on expression of target genes in individual cells within the animal using a standard 405 nm microscope-mounted laser to deliver the activating light. Activation by laser requires illumination times of <10 ms per cell. Thus, we have developed a straightforward and efficient tool to precisely control gene expression in the multicellular organism C. elegans.
|
|
Scooped by
mhryu@live.com
March 26, 4:13 PM
|
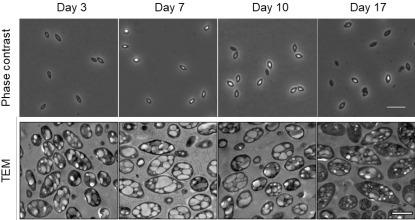
Heterotrophic marine bacteria frequently experience fluctuations in carbon availability driven by phytoplankton dynamics. As a result, bacteria undergo repeated cycles of rapid growth during brief resource pulses followed by prolonged starvation. Yet the mechanisms that support bacterial survival during nutrient limitation remain poorly understood. Here, we investigate starvation survival in the algal-associated bacterium Phaeobacter inhibens. We show that cells remain viable for extended periods under carbon depletion while undergoing physiological and morphological changes. Using electron microscopy, metabolomics, and genetic approaches, we identify intracellular polyhydroxybutyrate (PHB) granules as a key factor supporting survival during starvation. PHB accumulates during growth and is progressively consumed under carbon limitation. Deletion of the PHB synthase gene (phaC) eliminates granule formation and reduces long-term viability. Comparative analyses show that the genetic capacity for PHB biosynthesis is widespread among members of the Roseobacter group, suggesting a conserved strategy among algal-associated bacteria. However, species lacking PHB also survive starvation, indicating that additional mechanisms contribute to persistence under nutrient limitation. Together, our results identify intracellular carbon storage as a central mechanism linking bacterial physiology to survival in fluctuating marine environments, and highlight the diversity of strategies shaping microbial community dynamics and carbon cycling in the ocean.
|
|
Scooped by
mhryu@live.com
March 26, 2:52 PM
|
Alternative pre-mRNA splicing (AS) and nonsense-mediated decay (NMD) are key RNA-based regulatory mechanisms in eukaryotic cells. Although NMD was initially identified as a quality-control pathway targeting aberrant transcripts, increasing evidence indicates that it frequently operates in concert with genetically programmed AS to regulate the expression output of protein-coding genes. Here, we describe AS-NMD mechanisms, highlighting their diverse functions across biological contexts. These include roles in maintaining cellular homeostasis, preventing premature expression of differentiation-specific genes, sharpening gene expression dynamics during development, and fine-tuning responses to physiological cues. We additionally summarize experimental approaches used to study AS-NMD and discuss possible evolutionary mechanisms underlying the acquisition of new regulatory AS-NMD events and their integration into gene regulatory networks. Overall, this review provides a unified perspective on AS-NMD as a widespread, multifaceted, and evolutionarily dynamic regulator of gene expression at the post-transcriptional level.
|
|
Scooped by
mhryu@live.com
March 26, 12:50 AM
|
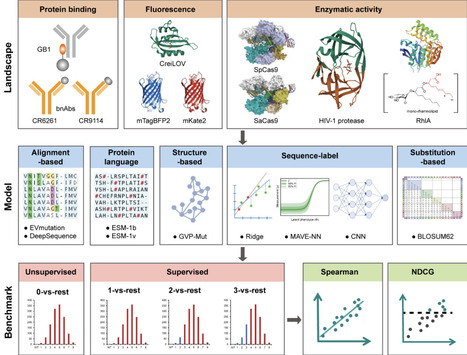
Combinatorial mutagenesis is essential for exploring protein sequence-function landscapes in engineering applications. However, while large-scale machine learning benchmarks exist for protein function prediction, they are primarily limited to single-mutant libraries, leaving a critical gap for combinatorial mutagenesis. Here we introduce CombinGym, a benchmarking platform featuring 14 curated combinatorial mutagenesis datasets spanning 9 proteins with diverse functional properties including binding affinity, fluorescence, and enzymatic activities. We evaluated nine machine learning algorithms from five methodological categories (alignment-based, protein language, structure-based, sequence-label, and substitution-based) across multiple prediction tasks, assessing both zero-shot and supervised learning performance using Spearman's ρ and Normalized Discounted Cumulative Gain metrics. Our analysis reveals the substantial impact of measurement noise and data processing strategies on model performance. By implementing hierarchical dataset splits (0-vs-rest, 1-vs-rest, 2-vs-rest, and 3-vs-rest scenarios), we demonstrate the value of lower-order mutation data for empowering machine learning models to predict higher-order mutant properties. We validated this capacity through both in silico simulation (improving fluorescence brightness of an oxygen-independent fluorescent protein) and experimental validation (engineering enzyme substrate specificity), achieving a substantial increase in specific activity. All datasets, benchmarks, and metrics are available through an interactive website (https://www.combingym.org), facilitating collaborative dataset expansion and model development through integration with automated biofoundry platforms.
|
|
Scooped by
mhryu@live.com
March 26, 12:43 AM
|
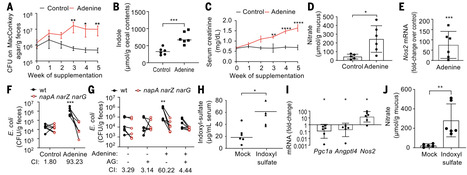
Chronic kidney disease (CKD) is linked to an elevated fecal abundance of Enterobacteriaceae, but the ecological drivers of this shift and its impact on disease progression remain unclear. The uremic toxin indoxyl sulfate is produced from microbiota-derived indole in the liver. Here, we found that in mice with adenine-induced CKD, impaired clearance of indoxyl sulfate elevated mucosal expression of the gene encoding inducible nitric oxide synthase (iNOS). The resulting rise in luminal nitrate levels promoted E. coli growth by means of nitrate respiration. Fecal microbiota from CKD patients generated more indole than feces of healthy controls during anaerobic culture, but only in the presence of nitrate. Nitrate enhanced indole production by E. coli, thereby worsening renal pathology in CKD mice, which was mitigated by iNOS inhibition.
|
|
Scooped by
mhryu@live.com
March 25, 11:49 PM
|
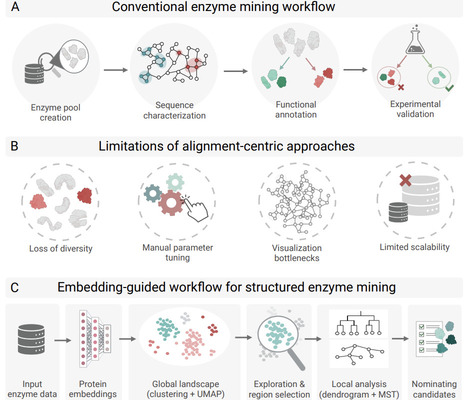
The rapid expansion of protein sequence databases continues to outpace functional characterization, limiting efficient enzyme discovery in large, heterogeneous, and sparsely annotated sequence spaces. Here, we present an embedding-guided framework for structured navigation of enzyme sequence space, implemented in SelectZyme. The workflow integrates protein language model embeddings with dimensionality reduction, hierarchical clustering, connectivity reconstruction, and quantitative dendrogram analysis to enable exploration without reliance on fixed sequence identity thresholds or predefined functional annotations. Across distinct case studies, we demonstrate that embedding-defined neighborhoods preserve fold-level coherence even when sequence identity resides in the twilight zone and that biologically meaningful functional organization can emerge under fully unsupervised conditions. In a large multi-family PETase landscape exceeding 100,000 sequences, the framework supports scalable, anchor-guided prioritization under sparse labeling and process-motivated constraints. By unifying latent representations with connectivity-aware and hierarchical interpretation, this approach reframes enzyme mining from threshold-driven similarity filtering into structured exploration, providing a scalable methodological foundation for hypothesisdriven biocatalyst discovery and reasonable starting points for downstream protein engineering.
|
|
Scooped by
mhryu@live.com
March 25, 11:03 PM
|
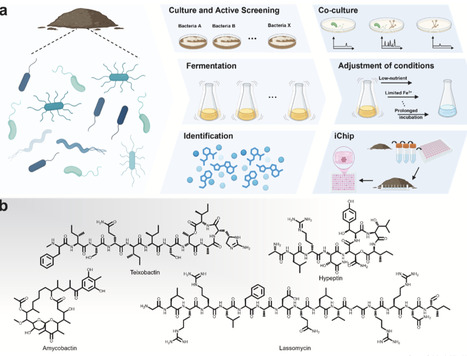
The discovery of antibiotics transformed modern medicine and extended the average human lifespan by decades. However, the initial golden era of antibiotic discovery has significantly waned, a decline aggravated by the relentless evolution of drug resistance among human pathogens, ultimately driving the current global antimicrobial resistance (AMR) crisis. The repeated rediscovery of known compounds from conventional soil-derived microbes underscores the urgent need for new strategies and ecological frontiers. Here, we review emerging directions in antibiotic discovery that collectively address this innovation gap. Refined cultivation techniques such as co-culture and iChip have reactivated rare taxa and yielded novel scaffolds like teixobactin. Multi-omics and synthetic biology approaches now enable culture-independent access to cryptic biosynthetic gene clusters from the vast uncultivated microbial majority. Most recently, artificial intelligence (AI) has expanded the search frontier to neglected taxa such as archaea and even evolutionary timeframes through paleoproteome mining. Overall, these innovations signal a new era of intelligent, data-driven antibiotic discovery. The integration of ecology, omics, synthetic biology, and AI provides a sustainable framework to replenish the antibiotic pipeline and mitigate the growing threat of AMR.
|
|
|
Scooped by
mhryu@live.com
March 26, 11:36 PM
|
Protein expression is an important aspect of synthetic biology, where host selection critically impacts the production efficiency. V. natriegens has emerged as a promising prokaryotic host due to its exceptionally rapid growth rate and broad substrate metabolic spectrum. Benefiting from the advances in annotation on microbial genomes and metabolic pathways, as well as the development of molecular biology tools, continuous progress has been made in bioproduction using V. natriegens. In this Perspective, we provide a systematic introduction of the basic biological properties of V. natriegens and present the engineered V. natriegens chassis which are currently used for protein expression. We summarize the latest synthetic biology components and genetic tools specifically designed for the modification of V. natriegens. Especially, we focus on the most recent study and application achievements of V. natriegens in in vivo and in vitro protein expression, which has received limited attention in previous reviews. This review aims to present researchers with a comprehensive overview of using V. natriegens for protein expression, highlight its advantages and application prospects, and present some challenges and future perspectives in this field, so as to promote wider research in the synthetic biology field.
|
|
Scooped by
mhryu@live.com
March 26, 9:17 PM
|
mRNA vaccines benefit from rapid design, scalable manufacturing and strong safety profiles, and they are being preclinically and clinically explored for various infectious diseases, cancer and autoimmune disorders. However, global access to mRNA vaccines remains limited by technical, logistical, economic, regulatory and ethical challenges. In this Review, we discuss the current mRNA vaccine landscape, from approved products to emerging candidates, and outline key barriers to equitable access. We highlight engineering strategies to address these barriers, including thermostability improvements, alternative administration routes, new delivery systems and alternative RNA platforms, such as self-amplifying and circular RNA, as well as the integration of artificial intelligence- and machine learning-based design and optimization. We also discuss the regulatory, ethical and social dimensions of vaccine deployment, as well as the importance of building public trust and community engagement. Coordinated technical and policy advances are essential to remove barriers, enhance access and advance global health equity. mRNA vaccines combine rapid design, scalable manufacturing and strong safety profiles with applications across infectious diseases, cancer and autoimmune disorders. However, global access to mRNA vaccines remains limited. This Review explores how achieving equitable access depends on overcoming technical, logistical, economic and regulatory barriers through innovations in thermostability, delivery systems, alternative RNA platforms, artificial intelligence-driven design and optimization, and coordinated policy strategies.
|
|
Scooped by
mhryu@live.com
March 26, 9:06 PM
|
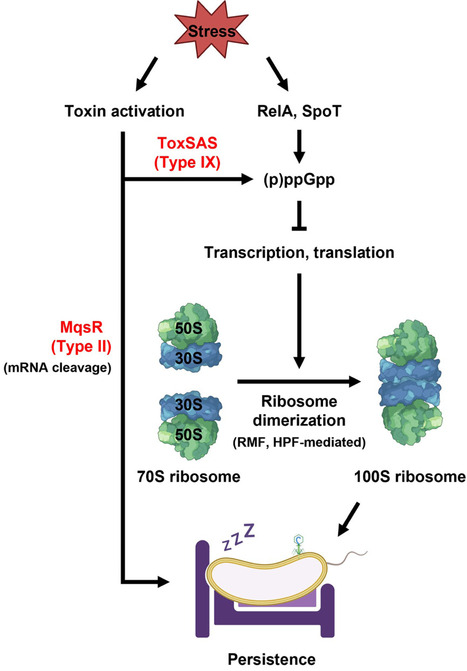
The toxin–antitoxin (TA) field continues to advance rapidly, especially given the recognition of TA systems as one of the most prevalent phage defenses. These systems are omnipresent in both bacteria and archaea and are currently classified into eight types. Here, we propose an additional type IX class, in which either the toxin or antitoxin utilizes nucleotide-based signaling. By establishing the new type IX TA system, the framework for the organization of TA systems, which is based on the diverse biochemical functions of both the toxins and antitoxins across the nine types, will be refined and aid in their discovery and understanding. In addition, we highlight the increasing evidence that TA systems are related to persistence and are activated by conformational changes in protein complexes.
|
|
Scooped by
mhryu@live.com
March 26, 8:43 PM
|
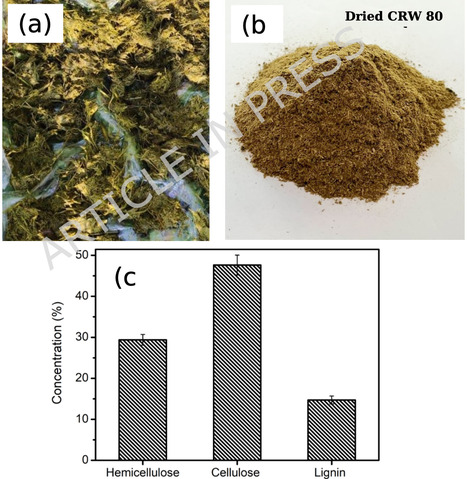
Cow rumen waste (CRW), a slaughterhouse by-product rich in lignocellulose, can be directly converted into bioethanol without harsh chemical pretreatment or additional microbial inocula. In this study, we developed an integrated enzymatic hydrolysis and fermentation process to produce bioethanol from CRW using Viscozyme Cassava CL and Saccharomyces cerevisiae. Key process parameters for enzymatic hydrolysis (enzyme concentration, pH, and substrate loading) were optimized, with 3 wt% enzyme at pH 5.0 and 50 g L− 1 CRW yielding the highest glucose release. The enzymatic hydrolysate was then fermented by S. cerevisiae under optimal conditions (inoculum 3 wt%, 30–35 °C, pH 5.0), producing an ethanol concentration of 4.79 g L− 1 with negligible residual sugar. These results demonstrate the feasibility of one-pot conversion of CRW to bioethanol. The novel enzyme-assisted process is scalable and low-cost, offering a sustainable strategy to valorize slaughterhouse waste into renewable bioethanol.
|
|
Scooped by
mhryu@live.com
March 26, 8:06 PM
|
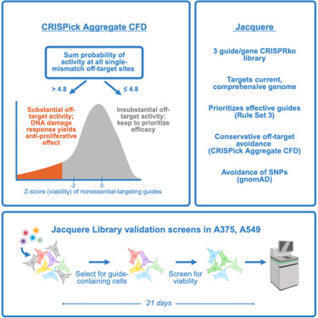
The continued development of high-dimensional CRISPR screen readouts, such as single-cell RNA sequencing and high-content imaging, necessitates compact libraries to enable functional interrogation at genome scale. Improved genome annotations cause library deprecation over time, further motivating an updated genome-wide design effort. Additionally, while on-target efficacy and off-target avoidance are often optimized in isolation, we lack a robust framework for simultaneously weighing and balancing these competing priorities. Here, we present a selection strategy that identifies guides with sufficient off-target activity to justify omission from the library, thus avoiding the unnecessary exclusion of active guides, allowing the inclusion of those with maximal on-target activity. We create, validate, and make available to the community the Jacquere library for knockout screens of the human genome, as well as its mouse counterpart, Julianna, to facilitate gene function discovery at scale.
|
|
Scooped by
mhryu@live.com
March 26, 7:35 PM
|
Current one-pot CRISPR diagnostics are limited by the cis-cleavage activity of Cas nucleases, which leads to amplicon degradation during amplification. Here, we report a streamlined strategy that overcomes this limitation. By integrating a bipartite split-crRNA into Cas12a (SCas12a), we separate target recognition from PAM dependency and completely eliminate cis-cleavage while preserving robust trans-cleavage. This strategy is broadly applicable for one-pot testing, compatible with recombinase polymerase amplification, RT–RPA, and loop-mediated isothermal amplification LAMP, as well as multiple Cas12a orthologs, including As, Lb, and Ct Cas12a. Moreover, the SCas12a accelerates one-pot testing with 100–1000-fold improved sensitivity and achieves >10-fold reduction in time-to-signal, enabling detection of targets at attomolar levels within 30 min. Additionally, it provides single-base resolution with up to 91-fold selectivity. The system has been successfully applied to detect HPV16, SARS-CoV-2, and TP53 SNPs in clinical samples. Together, we have developed a PAM-independent and cis-cleavage-free one-pot Cas12a assay, which holds strong potential for point-of-care diagnostics.
|
|
Scooped by
mhryu@live.com
March 26, 4:17 PM
|
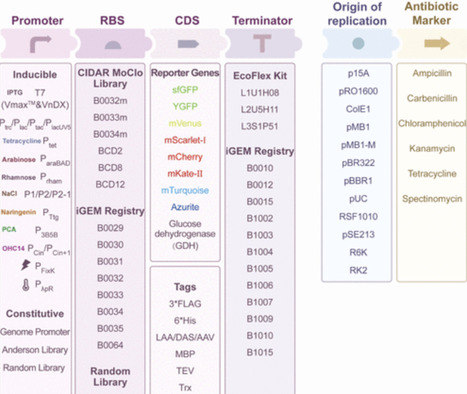
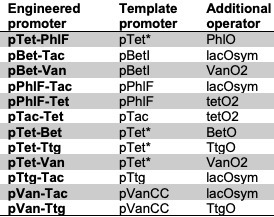
Engineering bacterial promoters to integrate multiple regulatory signals remains a formidable challenge. Juxtaposing operator sites frequently increases basal leakiness, compresses the fully induced state, and introduces severe sequence-context dependencies. Here, we systematically engineered two-input combinatorial promoters in E. coli that integrate signals from multiple transcription factors. To achieve precise operational control over these regulators, we drove the promoters using highly optimised, small-molecule-responsive sensors from the Marionette transcription-factor cassette, allowing us to assemble 19 reporter-specific, four-state truth tables across 12 distinct promoter architectures. We evaluated each design against a stringent statistical criterion for inducer-conditioned coincidence responses. Nine architectures satisfied this criterion, yielding a robust set of operational AND switches. By comparing successful and unsuccessful designs, we reveal that performance hinges primarily on suppressing partially induced states, ensuring structural compatibility between the promoter scaffold and the inserted operator, and precisely managing the orientation of long operators to avoid recreating unintended promoter-like motifs. Furthermore, reciprocal architectures and alternate downstream reporters frequently display divergent behaviors, underscoring profound asymmetries and local genetic-context dependencies. Ultimately, these findings deliver versatile combinatorial switches alongside practical, sequence-aware design rules for engineering multi-input bacterial promoters.
|
|
Scooped by
mhryu@live.com
March 26, 2:54 PM
|
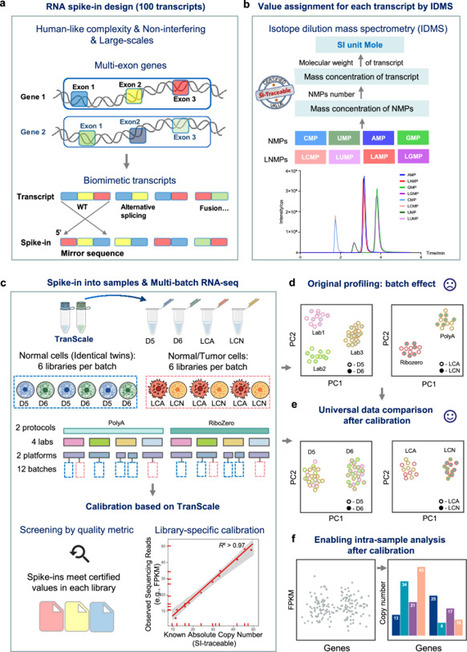
RNA-sequencing’s conversion of molecules to reads is inconsistent. Experiment-to-experiment variations (systemic bias) create batch effects, while gene-to-gene variations (sequence-dependent bias) invalidate inter-gene comparisons, precluding a universal scale. This confines analysis to relative fold-changes, a metric unreliable across batches. We introduce TranScale: 100 biomimetic standards with SI-traceable concentrations certified by Isotope Dilution Mass Spectrometry. Co-processed within samples, they empirically characterize systemic and sequence-dependent biases, generating a library-specific calibration curve (R² > 0.97) to convert reads into absolute quantities. This approach reveals that consistent fold-changes can mask severe absolute errors, exposing systemic biases missed by conventional QC. Across laboratories, this calibration reduced median inter-lab CV from >85% to <25% and increased biological signal-to-noise from ~0 to >7.9, outperforming the widely-used tool ComBat. By anchoring RNA-seq to the SI, our work establishes the metrological foundation for data interoperability and universal benchmarks, enabling absolute comparisons of SI-traceable quantities between any two genes. RNA-sequencing’s conversion of molecules to reads confines analysis to relative fold-changes, a metric unreliable across batches. Here, the authors introduce TranScale, a spike-in method with 100 biomimetic standards that converts reads into absolute quantities to resolve systemic biases.
|
|
Scooped by
mhryu@live.com
March 26, 2:01 PM
|
The bacterial pathogen Acinetobacter baumannii forms a resilient biofilm, enhancing bacterial survival in hostile environments. Although biofilm structure is primarily built from polysaccharides and nucleic acids, specific proteins are often essential for its development and integrity. Here, we identify microcins as a driver of biofilm formation in A. baumannii. Microcins are small (<10 kDa), secreted, antimicrobial proteins that typically mediate bacterial competition, yet their function beyond the antibacterial is unclear. We demonstrate that A. baumannii microcins use conserved sequence motifs to self-assemble into amyloid fibers, promoting biofilm formation. Scanning electron microscopy reveals microcin fibers extending from A. baumannii, suggesting a structural role as a biofilm scaffold. We also identify proteins required for microcin’s antibacterial function that are dispensable for biofilm formation, further distinguishing these two activities. Our findings show that microcins directly promote biofilm development and broaden understanding of the diverse functions of these understudied proteins.
|
|
Scooped by
mhryu@live.com
March 26, 12:46 AM
|
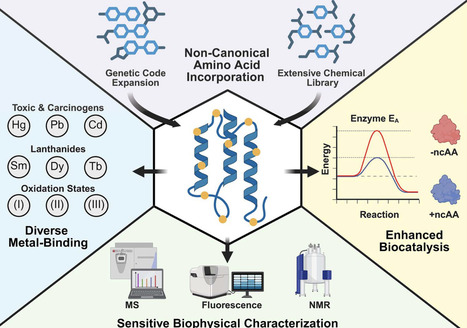
Non-canonical amino acids (ncAAs) are versatile molecular building blocks that can enhance nearly every aspect of protein engineering, from improving binding affinity to enabling precise quantitative analyses. This review highlights advances from 2020–2025 that demonstrate how expanding the amino acid repertoire unlocks new functionalities. We examine how genetically encoded ncAAs diversify and tune metal-binding properties, enabling programmable coordination and redox behavior in engineered enzymes. Further, we explore biocatalytic applications, including multi-fold activity enhancements in natural enzymes and the introduction of entirely novel reactivity in artificial systems. Finally, we discuss the growing use of ncAAs as intrinsic biophysical reporters, which support a wide range of spectroscopic methods for tracking structure, dynamics, and interactions at residue-level resolution. These capabilities establish ncAAs as essential tools that can be deployed at any stage of the protein design process, from constructing new catalytic centers to quantifying molecular behaviors in real time.
|
|
Scooped by
mhryu@live.com
March 25, 11:51 PM
|
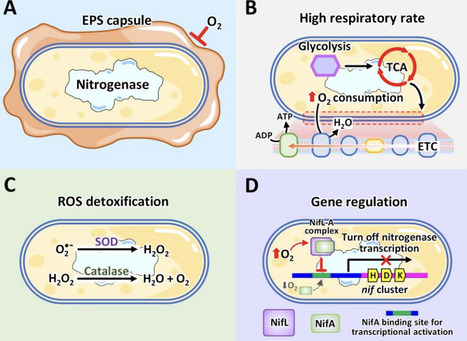
Ammonia (NH₃) is a critical molecule for agriculture, industry, and emerging energy applications. However, current industrial production by the Haber–Bosch process is energy-intensive and environmentally damaging, accounting for a significant portion of global CO₂ emissions. In contrast, biological nitrogen fixation (BNF) offers a sustainable alternative by converting atmospheric nitrogen (N₂) into NH₃ under ambient conditions through the action of nitrogenase enzymes. Among diazotrophic microorganisms, Azotobacter vinelandii stands out due to its ability to fix nitrogen aerobically, supported by unique physiological and genetic adaptations that protect its oxygen-sensitive nitrogenase. This review presents a comprehensive overview of NH₃ synthesis with A. vinelandii, highlighting its biochemical mechanisms, nitrogenase structure and function, and the protective strategies that enable aerobic nitrogen fixation. We further examine the diversity and advantages of Azotobacter strains, with a focus on A. vinelandii’s potential for engineered NH₃ production through synthetic biology, metabolic engineering, and emerging bio-based technologies such as photobiocatalysis and bioelectrochemistry. Recent innovations aimed at improving nitrogenase expression, cellular stability, and overall system efficiency are discussed in the context of advancing A. vinelandii as a robust chassis for industrially scalable, carbon-neutral NH₃ synthesis. The review emphasizes the importance of free-living nitrogen fixers in addressing the challenges of sustainable NH₃ production and provides insights into future directions for research and application.
|
|
Scooped by
mhryu@live.com
March 25, 11:24 PM
|
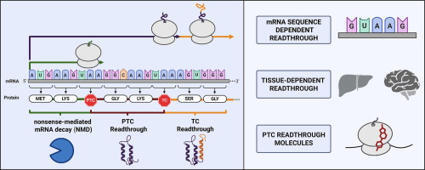
Accurate translation termination is essential for proteome integrity and is primarily governed by the release factors eRF1 and eRF3, which ensure precise recognition of stop codons and efficient release of nascent polypeptides. However, proteome integrity is challenged by mutations that generate premature termination codons (PTCs), leading to truncated, nonfunctional proteins and degradation of the aberrant transcript via nonsense-mediated mRNA decay (NMD). Collectively, these events account for ∼1800 human genetic diseases. Translational readthrough, the process by which near-cognate tRNAs decode stop codons and allow ribosomes to continue elongation beyond the stop codon, represents a possibility to suppress PTCs and restore full-length protein synthesis. Initially discovered in viruses as a mechanism to expand coding capacity, readthrough is now recognized as a regulated feature of eukaryotic gene expression influenced by both cis-acting sequence elements and trans-acting factors. Recent evidence highlights the remarkable context dependence of readthrough, revealing variation across transcripts, tissues, and developmental stages. In this review, we examine the molecular determinants that define stop codon recognition and readthrough efficiency, with particular emphasis on nucleotide context. We further discuss the mechanisms and binding sites of small molecules that promote PTC readthrough, and summarize the clinical development landscape of readthrough-inducing compounds for the treatment of diseases caused by nonsense mutations.
|