Your new post is loading...

|
Scooped by
mhryu@live.com
Today, 6:03 PM
|
To meet the needs of a growing human population, agricultural management practices have undergone substantial intensification, specialization and industrialization. This has contributed to biotic homogenization and a loss of diversity in microbial communities within agricultural systems. In this Perspective, we summarize recent studies that report microbial homogenization due to agricultural intensification. We propose a definition of microbial homogenization and explore how intensive agricultural practices can cause taxonomic, physiological, genetic and functional homogenization of microbial communities. Our analysis indicates that globally the diversity of rare taxa is lower in intensively managed agricultural lands compared with less-intensive lands and that agricultural intensification suppresses beneficial microorganisms and promotes pathogenic taxa. We identify microbial taxa that are sensitive to intensification and discuss how the disproportionate impact on rare microbiota can threaten agro-ecosystem functions and food security. Finally, we outline key challenges and suggest areas that require further research. In this Perspective, Banerjee, Dasgupta and van der Heijden discuss how intensive agricultural practices can lead to the taxonomic, physiological, genetic and functional homogenization of microbial communities and disproportionately affect rare microbiota.
|
|
Scooped by
mhryu@live.com
Today, 5:49 PM
|
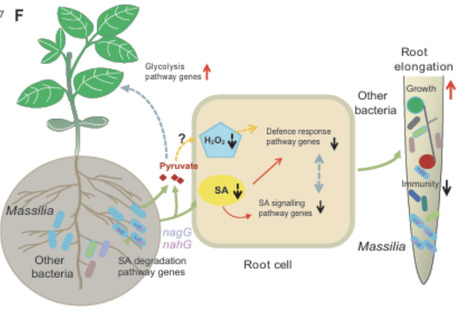
Rhizosphere microbiota play an important role in maintaining plant root growth. However, the physiological and molecular basis of microbial regulation of root traits remains poorly understood. Here, we report that Massilia efficiently colonizes roots and promotes root elongation. In particular, the M117 strain of Massilia significantly inhibits salicylic acid (SA)-related immune signalling, which is required for M117-mediated root elongation. M117 can directly degrade SA, thereby reducing SA levels in the roots. Integrated omics reveal the presence of multiple SA hydrolytic pathways in M117. Among them, the NagGHAaAb pathway is strongly induced by SA. This pathway regulates root growth and is nonrandomly distributed across Massilia species. Finally, we show that M117 colonization enriches specific bacterial taxa within roots. Our findings reveal a specific pathway employed by rhizosphere bacteria to colonize roots and promote their growth and highlight a useful microbial strategy and information for balancing host immunity and growth.
|
|
Scooped by
mhryu@live.com
Today, 5:36 PM
|
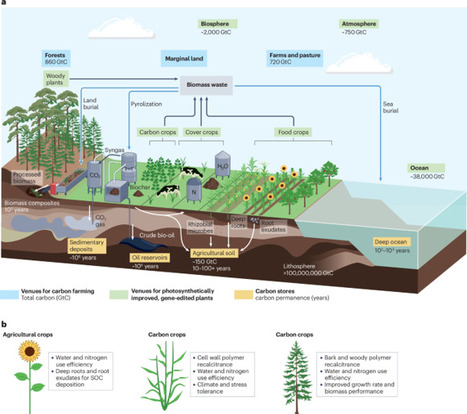
Anthropogenic carbon emissions have destabilized Earth’s carbon cycle, triggering cascading effects on climate and biodiversity. Plant-based carbon dioxide removal (CDR) presents a scalable, economically viable path to atmospheric carbon sequestration through soil carbon deposition, dedicated biomass cultivation and strategic agroforestry. Although photosynthesis drives terrestrial carbon capture, effective CDR strategies demand genetic optimization of carbon assimilation, retention and storage. The regulatory landscape is restrictive towards transgenic crops yet permissive of genome editing, creating a window for intervention. Advances in CRISPR-based editing, computational plant trait prediction and delivery systems for gene-editing tools in planta enable precision engineering of plant phenotypes to increase photosynthetic efficiency and carbon sequestration capacity. In this Review, we map the molecular and physiological innovations required to realize plant-based CDR at climate-relevant scales. Beyond optimizing carbon capture itself, we examine strategies to engineer enhanced biomass accumulation, improve nitrogen and water use efficiency, and stabilize carbon storage in plant and soil systems. We further assess the opportunities, implementation challenges and the potential of deploying genome-edited crops as a cornerstone of global carbon management. Gene editing to enhance photosynthesis in crop plants offers a strategy to boost plant carbon capture and mitigate climate change. This Review explores the agronomic, molecular and technical challenges in engineering photosynthesis for carbon sequestration as well as the genetic tools and editing technologies that can improve plant productivity and carbon storage.
|
|
Scooped by
mhryu@live.com
Today, 5:25 PM
|
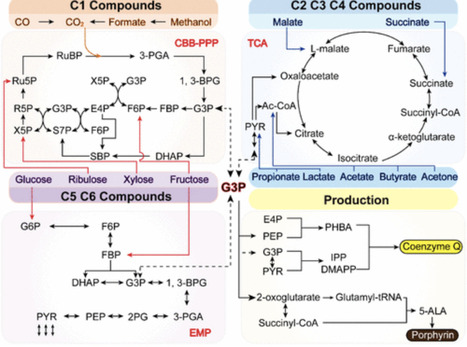
Rhodobacter sphaeroides, a purple nonsulfur photosynthetic bacterium, displays exceptional metabolic versatility, enabling growth under both aerobic and anaerobic conditions and utilization of diverse carbon sources. Its flexible metabolism, combined with native pathways for terpenoid and tetrapyrrole biosynthesis, makes it a highly promising microbial chassis for the production of valuable compounds. Advances in metabolic and synthetic biology have allowed the engineering of R. sphaeroides for the efficient synthesis of coenzyme Q10 (CoQ10) and porphyrin derivatives through strategies such as precursor supply enhancement, pathway optimization, modulation of redox and energy balance, manipulation of global regulatory systems, and fermentation control. Beyond CoQ10 and porphyrins, this organism holds the potential to produce hydrogen, carotenoids, and other high-value terpenoids. This review summarizes the metabolic features, native regulatory networks, and engineering approaches in R. sphaeroides, highlighting its versatility and robustness as a platform organism. The insights provided here underscore its potential as a chassis for synthetic biology applications and industrial bioproduction of a wide range of bioactive compounds.
|
|
Scooped by
mhryu@live.com
Today, 5:18 PM
|
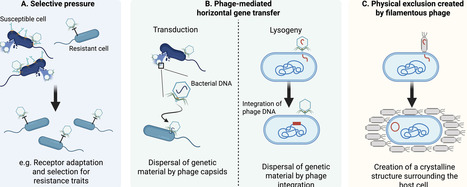
Microbial communities deliver essential functions in ecosystems. In plant environments, the plant microbiome facilitates nutrient uptake, supports plants during abiotic stress, and counteracts disease. As implementation of synthetic microbial communities becomes more of a realistic strategy for mitigating the effects of biotic and abiotic stressors on plant productivity, it is increasingly important to understand how interactions between microbes, which are essential for ecosystem function (hub microbes), are maintained. Recent research highlights the ecological role of bacteriophages, the viruses of bacteria, in host-associated microbial communities. Current evidence demonstrates the influence of the phageome on microbiomes, ranging from effects on an individual (transduction, lysogenic conversion, and evolutionary pressure) to entire populations and communities, such as Kill-the-Winner dynamics. These dynamics appear to affect the overall function of microbial communities and support plant growth. In this review, we lay out recent insights on the role of bacteriophages in plant-associated microbiomes through an eco-evolutionary lens and future directions of research to broaden our understanding of the ecological implications of bacteriophages.
|
|
Scooped by
mhryu@live.com
Today, 5:09 PM
|
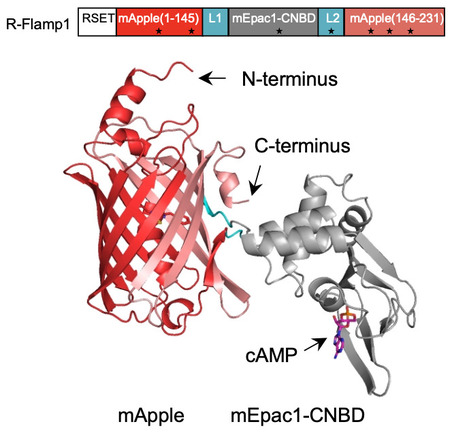
Circularly permuted green fluorescent protein (cpGFP)-based high-performance cAMP sensors have enabled real-time monitoring of cAMP dynamics with high spatiotemporal resolution in living animals. However, their utility is hampered by significant spectral overlap with other green/yellow fluorescent indicators and blue/cyan light-activated optogenetic actuators, limiting their compatibility in multiplexed imaging applications. While existing red cAMP sensors offer great spectral separation, they often suffer from a limited dynamic range ( < 1.5-fold in HEK293T cells), low cellular brightness, aggregation, or significant blue-light-induced photoactivation. Here, we report R-Flamp1, a red cAMP sensor with a large dynamic range ( > 10-fold in HEK293T cells), enhanced cellular brightness, appropriate cAMP affinity (Kd ~1.9 μM), subsecond response kinetics, and minimal photoactivation under blue or cyan light exposure. Using R-Flamp1, we visualized region-specific cAMP dynamics, and when paired with green indicators, revealed differential activation patterns between cAMP and neuromodulators or calcium using two-photon imaging and fiber photometry during various behaviors. These findings provide valuable insights into the role of cAMP signaling in complex behaviors. R-Flamp1 is a high-performance red fluorescent cAMP sensor and the authors monitor region-specific cAMP dynamics in vivo and reveal distinct activation patterns between cAMP, calcium, and neuromodulators via two-photon imaging and fiber photometry in animals.
|
|
Scooped by
mhryu@live.com
Today, 3:47 PM
|
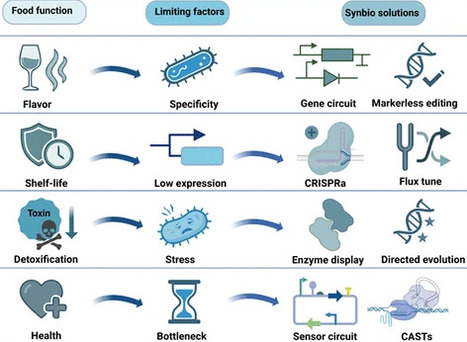
Lactic acid bacteria (LAB), as one of the key microorganisms in the food industry, play a crucial role in functional food fermentation. However, current LAB strains are limited by challenges, such as plasmid instability, low gene expression efficiency, and complex regulation of metabolic fluxes, which hinder their broader application. This review provides an overview of the traditional applications of LAB while highlighting current limitations that constrain their effective use. Then, it focuses on synthetic biology-driven strategies for precisely designing and expanding functions through gene editing, metabolic engineering, and genetic circuits. Finally, this review discusses how to improve gene expression efficiency in LAB and the use of directed evolution to optimize exogenous genes, offering perspectives for future research in the development of personalized food applications. The emerging tools of synthetic biology will further improve the production efficiency and product diversity and meet the needs of consumers for high-quality, multifunctional, and personalized food.
|
|
Scooped by
mhryu@live.com
Today, 1:37 AM
|
The endosymbiotic evolution of plastids and mitochondria was central to the origin and success of eukaryotes. One of the most prominent molecular machineries thought to have disappeared early in eukaryote evolution is the multi-subunit bacterial DNA polymerase III (DNApol-III), which is the principal enzyme complex supporting DNA replication in bacteria. Here, we combined worldwide metagenomics and cultivation to characterisz the mosaic genomic landscape of abundant phytoplankton lineages of Teleaulax (Cryptophyceae), which contain an endosymbiotically-derived nucleomorph genome. Unexpectedly, the nuclear, plastid and nucleomorph genomes of Teleaulax contain ubiquitously expressed genes for plastid-targeted DNApol-III subunits. These genes shed light on the functioning of Teleaulax genomes when sequestered by the ciliate Mesodinium during its kleptoplastidic photosynthetic activity. In particular, the alpha subunit gene (encoding the polymerase activity), which resides in the nucleomorph genome, is continuously expressed in Mesodinium in controlled laboratory experiments. This provides a mechanistic explanation for the replication of Teleaulax plastid genomes weeks after the nuclear genome is lost. Beyond Teleaulax and close relatives, we also identified genes encoding plastid-targeted DNApol-III subunits (including alpha) in nuclear genomes of unicellular and multicellular lineages of Archaeplastida that form, along with those of Cryptophyceae, monophyletic clades firmly positioned within Cyanobacteria. Together, our results reveal a previously overlooked retention of bacterial DNA replication machinery from plastid primary endosymbiosis in Archaeplastida, its acquisition by Cryptophyceae during secondary endosymbiosis, and its direct role in contemporary plankton as a facilitator of kleptoplastidic photosynthetic activity by heterotrophic ciliates.
|
|
Scooped by
mhryu@live.com
Today, 1:26 AM
|
Bacteriophages (phages) play essential roles in microbial systems, yet most phage proteins remain poorly characterized. Protein tertiary and quaternary structure information contributes valuable information about protein function. As many phage proteins function as homooligomers, complexes that consist of multiple identical subunits, there is great interest in computationally predicting their configurations. Here we present a computational framework, the Phage Homomer Level Estimate and Generation Method (PHLEGM) for inferring homooligomeric states directly from the protein sequence by combining AlphaFold-Multimer modelling with inter-subunit interface quality assessment. We proceeded to experimentally validate two out of nine predicted homooligomers using size exclusion chromatography and complementary hydrodynamic techniques. These efforts confirmed our predictions for a dimer and a trimer, highlighting the value of experimentally benchmarked computational predictions and showing the challenges of heterologous phage protein production. Applied to >22,000 phage protein sequences in the PHROGs database, our approach revealed extensive diversity in phage homooligomeric protein complexes. Benchmarking against protein language model-based predictors on a curated reference set of known phage homooligomers demonstrated superior accuracy of our structure-based method, achieving robust performance in classifying protein homooligomeric states, with the highest accuracy observed for trimers and higher-order complexes. These results highlight the value of computational predictions to decipher the complexities of the vast viral sequence space. All predicted complex structures and functional inferences are made publicly available to support structural and functional studies of phage proteins.
|
|
Scooped by
mhryu@live.com
Today, 12:45 AM
|
Cross-feeding interactions, in which a producer species release by-products that serve as resources for a consumer species, play an important role in shaping microbial community diversity. Producers create opportunities for consumers by supplying high-energy resources that are often scarce in the environment. However, they also exert strong top-down effects by releasing metabolites in pulses and generating spatial gradients of resource availability. How these spatiotemporal constraints shape consumer evolution remains poorly understood. To address this question, we used a two-species cross-feeding system in which Acinetobacter johnsonii excretes benzoate (a by-product of benzyl alcohol oxidation) into the external environment where it is consumed by Pseudomonas putida. To assess how the origin of benzoate (externally supplied or produced by cross-feeding) shapes consumer evolution, we evolved P. putida for 200 generations in monoculture or in co-culture with A. johnsonii. Populations evolved in monoculture exhibited improved growth relative to the ancestor, whereas populations evolved under cross-feeding showed little to no growth improvement. Whole-genome sequencing revealed pervasive loss-of-function mutations in flagellar genes among populations evolved in monoculture, but not under cross-feeding conditions. High-throughput imaging assays showed that populations evolved under cross-feeding not only maintained but also enhanced functional motility. Competition experiments with single mutants revealed context-dependent fitness effects: loss-of-function mutations were highly beneficial when benzoate was externally supplied but deleterious when benzoate was supplied by A. johnsonii, highlighting the importance of motility in cross-feeding interactions. Together, our results show that resource origin fundamentally reshapes selective pressures and alters evolutionary outcomes in microbial communities.
|
|
Scooped by
mhryu@live.com
Today, 12:18 AM
|
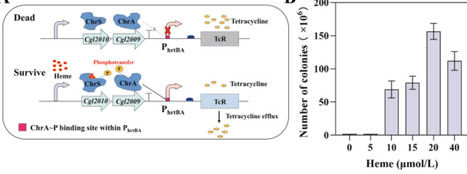
Heme is an essential cofactor involved in diverse cellular processes and is an important target for microbial biosynthesis. However, engineering microbial heme production remains challenging due to the requirement for coordinated activity of multiple pathway enzymes and the lack of scalable strategies for enzyme-level screening. In this study, we developed a growth-coupled screening system in Corynebacterium glutamicum by establishing a ChrSA-based heme sensor derived from the native heme-responsive two-component system, which directly converts intracellular heme levels into a selectable growth phenotype. Using this biosensor-enabled system, random mutagenesis libraries were constructed for four key enzymes of the coproporphyrin-dependent (CPD) heme biosynthesis pathway, and improved variants were identified through biosensor-guided selection. The selected variants were subsequently validated by chromosomal allelic replacement, resulting in an overall increase in heme production of approximately 38.9% and a final titer of 308.7 mg/L under native, single-copy regulation. Sequence and structural analyses indicated that the beneficial substitutions were predominantly located outside catalytic residues, suggesting that changes in enzyme stability and conformational properties contributed to the enhanced heme biosynthesis. This work established a heme biosensor-guided strategy for enzyme variant screening in tightly coupled metabolic pathways and provided a practical systems-level approach for microbial heme pathway engineering.
|
|
Scooped by
mhryu@live.com
Today, 12:11 AM
|
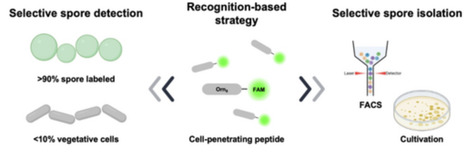
We identify a peptide discovered through fluorescence-based screening of a cell-penetrating peptide (CPP) library that preferentially associates with bacterial spores and evaluate its utility as a fluorescence-guided tool for spore isolation. Specifically, an eight-ornithine peptide conjugated with 5(6)-carboxyfluorescein (Orn8-K-FAM) generates a distinct and spatially confined fluorescence signal in spores. Across multiple spore-forming bacterial species, more than 90% of spores were labelled by Orn8-K-FAM, whereas only a minor fraction of vegetative cells from non-spore-forming bacteria exhibited detectable fluorescence, indicating a high degree of preference at the cellular level under the conditions tested. To assess practical applicability, Orn8-K-FAM labeling was combined with flow-cytometric sorting FACS, enabling fluorescence-guided isolation of spores from mixed microbial populations, including both a defined synthetic community and an environmentally derived bacterial fraction. Compared with conventional spore isolation approaches based on selective cultivation or heat-mediated inactivation of vegetative cells, this workflow provides a cultivation-independent means to separate spore-associated cellular states from heterogeneous microbial samples. This work highlights the potential of CPP-derived probes as analytical tools for spore-focused studies in applied and environmental microbiology, while providing a foundation for further investigation into the scope and mechanistic basis of CPP-spore interactions.
|
|
Scooped by
mhryu@live.com
Today, 12:03 AM
|
Human-associated microbial genomes encode extensive strain-level diversity and niche-specific gene repertoires that are critical to host health. However, these complex sequence features remain difficult to capture using general-purpose DNA foundation models, highlighting the need for dedicated representation learning tailored to the human microbiome. Here, we introduce Genos-m, an open-source foundation model for human-associated microbial genome representation. Genos-m was pretrained on approximately 1.2 trillion nucleotide tokens from a curated microbial genome corpus, including human-associated prokaryotic isolates, high-quality metagenome-assembled genomes (MAGs) and bacteriophages, supplemented with GTDB species-level representative genomes to broaden prokaryotic taxonomic breadth. The model uses a sparsely activated Mixture-of-Experts (MoE) Transformer architecture, with 4.7 billion total parameters, approximately 330 million activated parameters per forward pass and a maximum context length of one million base pairs. We evaluated frozen Genos-m representations across short-sequence and gene-level tasks, biosynthetic gene cluster (BGC)-based regional sequence tasks, whole-genome strain phenotype prediction, and zero-shot transfer on prokaryote-related RNAfitness assays. Across these benchmarks, Genos-m consistently ranked among the leading comparison models, with the best performance in five of eight gene-fitness regression tasks and in BGC type classification. Using sparse autoencoders, we identified sparse features in Genos-m hidden activations that aligned with annotated ORFs, intergenic regions, and tRNA and rRNA loci. In downstream applications, Genos-m-derived genome-informed species representations incorporated into a human microbiome self-supervised learning model improved colorectal cancer (CRC)-control classification over conventional species-abundance random forest models. Genos-m also generated stable sample-level embeddings from as few as 10,000 metagenomic reads, retaining gut microbial community structure that distinguished geographic origin and aligned with enterotypes defined from full-depth taxonomic profiles. Together, these results support Genos-m as a reusable representation model for microbial genomes and metagenomes, with conclusions bounded by the reported datasets, task definitions and evaluation protocols. Genos-m model weights, inference code, and usage documentation are publicly available on GitHub (https://github.com/BGI-HangzhouAI/Genos-m) and HuggingFace (https://huggingface.co/BGI-HangzhouAI/Genos-m).
|
|
|
Scooped by
mhryu@live.com
Today, 5:55 PM
|
Plasmids drive evolution by transferring traits across microbial hosts. Transmission depends on both host–plasmid (infection) and plasmid–plasmid (compatibility) interactions, yet how the structure of these networks shapes transmission remains poorly understood. We hypothesized that these two ecological networks interact in non-additive ways to influence community outcomes. To test this, we developed a stochastic agent-based model that embeds both network structures and simulates coupled host–plasmid dynamics. We systematically varied the structure of each network, both individually and in combination, to isolate the effect of structure on host-plasmid dynamics. A modular (interactions organized into clusters) and hub (interactions concentrated on the highly connected) plasmid-plasmid compatibility network promoted transient host coexistence, while a modular host-plasmid infection network promoted plasmid diversity and stable host coexistence. Importantly, structured networks interacted non-additively, and their impact was most apparent when plasmid carriage imposed a moderate fitness cost on hosts. For example, combining a modular infection network with a hub compatibility network reversed the expected plasmid prevalence patterns, demonstrating that the structure of one network can counteract the effects of the other. We further re-parameterized our model to recapitulate empirical host-plasmid community dynamics, showing that infection network structure can strongly shape plasmid prevalence even in the presence of substantial biological heterogeneity. Our results highlight the necessity of jointly considering host–plasmid infection and plasmid–plasmid compatibility networks to understand host–plasmid community dynamics and their eco-evolutionary potential. More broadly, this work provides an initial mechanistic framework for generating testable hypotheses and underscores that systems involving multiple hosts and infectious agents require explicit consideration of how different ecological networks interact to shape community dynamics.
|
|
Scooped by
mhryu@live.com
Today, 5:46 PM
|
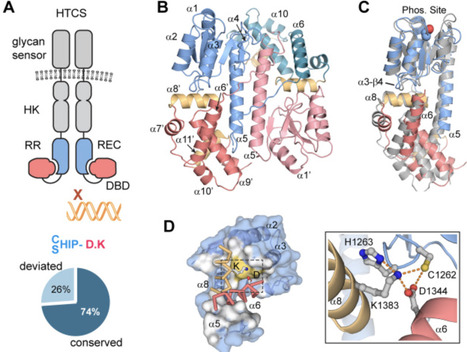
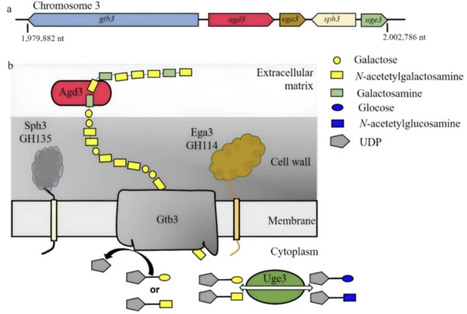
Human gut microbes, such as Bacteroides, rely on specialized gene clusters known as polysaccharide utilization loci (PULs) to metabolize diverse dietary and host-derived glycans. A major class of transcription regulators of these PULs is the hybrid two-component system (HTCS) containing a histidine sensor kinase and a response regulator (RR) within a single transmembrane polypeptide chain. Characterizing HTCS-mediated PUL regulation is often challenging because the specific glycan signals required to activate most HTCSs remain unknown. Here, we characterized structural details of a highly conserved HTCS activation mechanism and developed a universal activation strategy by mutating the interdomain latch motif that inhibits the DNA-binding activities. Using the RR portion of BT4124 from Bacteroides thetaiotaomicron as a model system, crystallographic analyses reveal a “closed” inactive conformation anchored by a hydrogen-bond network formed by the conserved latch residues between the receiver and DNA-binding domains. Molecular dynamic simulation with the deep-learning BioEmu shows that the “AD” mutation of the latch residues destabilizes the inhibitory interface, shifting the conformation equilibrium predominantly to an active, “open” conformation. This constitutively active variant, BT4124RAD, allows us to map specific DNA-binding sites within the potential regulated promoters in vitro and characterize transcription regulation in cells. Induced expression of BT4124RAD not only down-regulates local homogalacturonan (HG) utilization genes but also cross-represses multiple PULs associated with other HG-related pectic glycans. These findings highlight a complex cross-regulatory network governing pectin degradation and establish the targeted latch mutation as a potential broadly applicable tool for deciphering the regulatory networks of HTCSs in Bacteroides.
|
|
Scooped by
mhryu@live.com
Today, 5:28 PM
|
Methanol has emerged as a sustainable C1 feedstock owing to its compatibility with existing infrastructure and the potential for renewable production from CO₂ and green hydrogen. Methylotrophic yeasts, including Komagataella phaffii (Pichia pastoris) and Ogataea polymorpha, can natively assimilate methanol and therefore represent attractive hosts for biomanufacturing. However, industrial application of methanol-based processes remains constrained by cytotoxicity, redox imbalance, and limited productivity compared to sugar-based fermentations. To address these challenges, extensive metabolic engineering strategies have been implemented to enhance methanol assimilation and redirect carbon flux toward value-added products. Over the past decade, remarkable progress has been achieved through the development of synthetic methylotrophy in non-methylotrophic yeasts, the expansion of product portfolios to glycans, fatty acid derivatives, polyketides, terpenoids, organic acids, and polyols, and the integration of multi-omics tools for systems-level design. This review summarizes recent advances in methanol assimilation enhancement, synthetic pathway construction, and fermentation engineering, highlighting strategies such as metabolic engineering and dynamic bioprocess control. In addition, current challenges and future perspectives are discussed with an emphasis on overcoming toxicity, improving efficiency, and establishing advanced methylotrophic yeasts as robust cell factories for sustainable C1-based biomanufacturing.
|
|
Scooped by
mhryu@live.com
Today, 5:21 PM
|
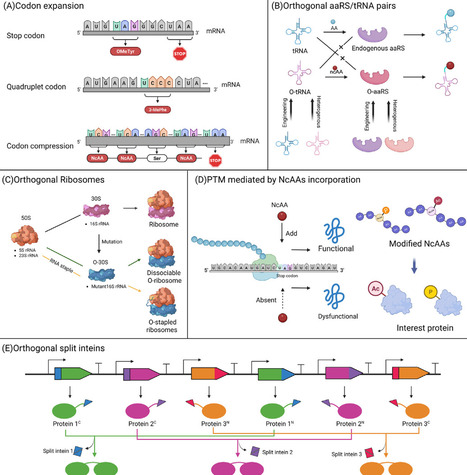
Precise regulation of protein abundance is essential for cellular function and physiology. Conventional approaches are often limited by insufficient resolution or unintended crosstalk. In contrast, orthogonal control technologies enable programmable and precise modulation of protein abundance while remaining insulated from native networks. In this review, we summarize the development and application of regulation technologies with different orthogonality across multiple levels. Orthogonal transcriptional control primarily involves the design and engineering of orthogonal RNA polymerases and transcription factors; orthogonal translational regulation focuses on advances in genetic codon expansion and post-translational modifications; targeted protein degradation and compartmentalized regulation are also discussed. Finally, we highlight the integration across the different levels described above. This review might bring disruptive insights and conceptual breakthroughs to precision medicine and sustainable biomanufacturing.
|
|
Scooped by
mhryu@live.com
Today, 5:16 PM
|
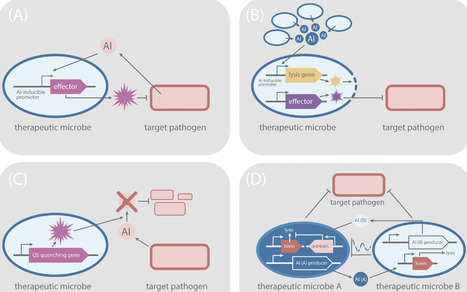
Quorum sensing (QS) is a cell–cell communication mechanism that enables bacteria to coordinate gene expression in response to population density and community composition. In many pathogens, QS plays a central role in host colonization and virulence, making it an attractive target for antimicrobial intervention. Synthetic biology offers powerful tools to exploit this vulnerability by either disrupting QS signaling or engineering microorganisms with QS-based circuits to detect and eliminate pathogens. In this review, we examine how QS and QS interference can be harnessed for QS circuit engineering and translated into applications such as therapeutic microorganisms. We also highlight the transition of QS research from fundamental microbiology to translational biotechnology, underscoring its potential to drive innovative strategies against microbial virulence and antimicrobial resistance.
|
|
Scooped by
mhryu@live.com
Today, 5:07 PM
|
Fungal exopolysaccharides (EPSs) are increasingly recognized as structurally programmable microbial polymers with applications spanning biomedicine, materials engineering, food systems, and environmental technologies. While previous reviews have often addressed fungal EPS diversity, production variables, or application domains separately, an integrated framework linking biosynthesis, molecular architecture, process control, and translational manufacturing remains underdeveloped. This review positions fungal EPSs as next-generation biomaterials by integrating (i) biochemical and genetic regulation of EPS biosynthesis, (ii) structure–function mapping across major polymer classes, (iii) cultivation and downstream processing workflows that enable reproducible product specifications, and (iv) industrial translation pathways within scalable and sustainability-aligned biomanufacturing systems. Gene-cluster–resolved case studies and process-to-product design principles illustrate how metabolic flux, fermentation parameters, and polymer modification shape functional performance. Current bottlenecks—including strain-dependent variability, purification complexity, quality harmonization, and techno-economic constraints—are critically evaluated to distinguish laboratory potential from scalable feasibility. By shifting from descriptive cataloging toward platform-based engineering logic, this review provides a translational roadmap for rational fungal EPS design within standardized and application-driven manufacturing frameworks.
|
|
Scooped by
mhryu@live.com
Today, 3:46 PM
|
The tricarboxylic acid (TCA) cycle is an essential part of the central metabolic hub that provides energy and biosynthetic precursors. Efficient regulation of central carbon flux is critical for maintaining optimal productivity of microbial cell factories (MCFs). However, biosensors capable of sensing TCA intermediates remain limited. Here, we engineered the catabolite control protein C (CcpC) from Bacillus species to reconstruct citrate-responsive biosensors in E. coli. Through hybrid promoter engineering, we systematically characterized and identified the functional roles of two CcpC binding sites. By applying the hybrid promoter, the engineered biosensor BcCcpC-PLBs exhibited the broadest dynamic range and highest expression level among its counterparts. Ligand profiling revealed the diverse responsiveness of BcCcpC to multiple metabolites of the TCA cycle. By structure-guided mutagenesis of BcCcpC, the obtained variant BcCcpC(S138L) exhibited an improved dynamic range of up to 3.02-fold under 80 mM citrate induction. This work establishes the first transcription factor (TF)-based citrate-responsive biosensor, which broadens the regulatory toolkit for central metabolism engineering.
|
|
Scooped by
mhryu@live.com
Today, 1:31 AM
|
DNA methylation plays critical roles in gene regulation in bacteria, from regulating essential processes like the cell cycle to phenotypes of practical interest like pathogenicity and motility. Synthetic manipulation of global methylation levels has broad impacts on cellular physiology, changing expression patterns of hundreds of genes. However, whether or how environmental variation in natural settings similarly impacts DNA methylation patterns has been unclear. In this work, using the alphaproteobacteria Methylobacterium extorquens and Caulobacter crescentus as model systems, we discover the methylome is highly fluid in response to environmental variation, with different environments leading to distinct patterns of increased or decreased methylation levels along the chromosome. Despite a heterogeneous effect of different environments on methylation patterns, we find a general principle where the dependence of methylation states on position in the genome decreases in proportion to growth rate. A simple model that considers the methylation state through different phases of the cell cycle as a function of distance from an origin provides a framework to interpret the effects of different stressors upon the observed environmental responsiveness of the methylation patterns. Our work highlights how sequencing data alone can shed light on important aspects of microbial physiology.
|
|
Scooped by
mhryu@live.com
Today, 1:24 AM
|
Plasmids frequently impose measurable fitness costs on their bacterial hosts, yet they remain abundant across clinical and environmental microbiomes. This apparent contradiction, known as the plasmid paradox, has traditionally been explained through mechanisms such as horizontal gene transfer, compensatory evolution, addiction systems, and fluctuating selection. Here we suggest that part of the paradox may arise from implicit physiological assumptions embedded in most empirical measurements—specifically, the assumption that growth rate is a direct proxy for fitness and that plasmid burden necessarily reduces it. We argue that these assumptions may not hold under many ecological conditions. We formalize cell division time as the maximum of several required cellular modules, including cytoplasmic biosynthesis and membrane or envelope synthesis. If plasmid carriage primarily increases cytoplasmic demand, its cost will be expressed only when cytoplasmic processes constitute the dominant bottleneck for growth. When other modules limit division, plasmid-associated burdens may be physiologically real yet evolutionarily silent. More broadly, equating fitness with maximal exponential growth rate overlooks well-established growth-survival trade-offs in bacteria, suggesting that plasmid costs measured under optimized laboratory conditions may systematically overestimate ecological selection against plasmid carriage.
|
|
Scooped by
mhryu@live.com
Today, 12:41 AM
|
Bottom-up manufacturing of structural DNA nanotechnology requires a long single-stranded DNA (ssDNA) scaffold and hundreds of short (~30 nt) ssDNA staples. However, scaling production remains bottlenecked by the high economic cost and environmental footprint of solid-phase chemical staple synthesis. To address these limitations, a phage-free, biological nanomanufacturing platform engineered in E. coli is developed here. Two intracellular strategies for producing programmable ssDNA were systematically evaluated: retron-based multicopy ssDNA (msDNA) synthesis via the Ec67 system and plasmid-encoded rolling circle replication (RCR). While native structural topology constraints within the retron (msd) cassette limit its sequence-design flexibility, the alternative RCR-driven engine successfully decouples ssDNA replication from sequence secondary structures, enabling the synthesis of arbitrary staples. This RCR platform reliably generates long circular ssDNA (cssDNA) precursors of at least 1.8 kb with exceptional sequence fidelity (>99%). Integrating programmable BseGI cleavage sites allows targeted strand-selective enzymatic processing to cleanly release stoichiometric, origami-grade pools of 32-nt staple strands. Atomic force microscopy (AFM) confirms that these biologically produced staples successfully drive the high-fidelity self-assembly of complex DNA tiles and hollow tubules. Crucially, robust structural folding is demonstrated directly within crude, unpurified cellular lysates, establishing a green, cost-effective framework for the one-pot fabrication of advanced DNA-based nanomaterials.
|
|
Scooped by
mhryu@live.com
Today, 12:15 AM
|
Climate change increasingly threatens global agriculture by intensifying abiotic stresses and destabilizing crop productivity, necessitating a deeper understanding of root-mediated traits governing resource acquisition and stress resilience. Here, we synthesize recent advances in root-centered plant phenomics, emphasizing how high-throughput phenotyping (HTP) enables high-resolution, scalable characterization of complex root traits and robust comparative analysis across diverse genotypes and environments. Innovations in multimodal imaging notably X-ray computed tomography (CT), magnetic resonance imaging (MRI), and machine learning-integrated rhizotrons facilitate detailed reconstruction of root system architecture and its temporal dynamics under both controlled and semi-field conditions. Furthermore, root phenotyping is increasingly interpreted within an integrated whole-plant framework. The integration of organ-specific assessments with physiological phenomics leveraging spectral and thermal data enables the characterization of developmental plasticity and root-mediated processes, including water-use dynamics, nutrient acquisition, and canopy stress responses under heterogeneous field conditions. These approaches link root traits such as rooting depth and spatial distribution to canopy-level physiological responses under stress. Despite these advances, significant bottlenecks persist in data interoperability, analytical scalability, and protocol standardization. Future progress will require integration of root phenomics with genomics, predictive modeling, and digital twin frameworks to improve resource-use efficiency, yield stability, and climate resilience in global cropping systems.
|
|
Scooped by
mhryu@live.com
Today, 12:04 AM
|
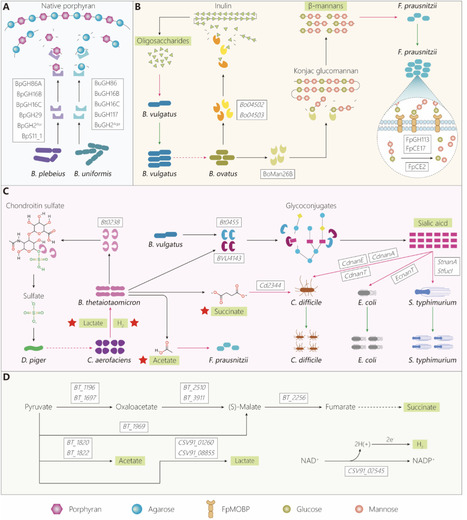
Methodological inconsistencies hinder the identification of gut keystone bacteria and their functional mechanisms. This work critically compares identification approaches, elucidates their interactions within the microbiota and host, and systematically details targeted dietary interventions. By explicitly linking keystone identification to diet-driven modulation, it provides a practical and precise framework for developing gut health strategies and managing microbiota-related diseases. review
|