 Your new post is loading...

|
Scooped by
mhryu@live.com
Today, 1:53 AM
|
Plant roots form a microbiome that interacts at the cell wall extracellular matrix before entering the cell. The root primary and accessory walls present a dynamic, cell-type-dependent scaffold that microbes must navigate, using shared cellulose or contrasting chitin motifs and influencing plant gene responses that encode enzymes for cell wall biosynthesis and degradation. We propose that an interface evolves as microbes reach the root tip and interact with host polymers, potentially driving concurrent degradation of root and microbial cells. Knowledge gaps span diffusion, fluid flow, nutrient exchange, and the physics of microbial motion within the wall boundary. Advances in in situ imaging and mathematical modelling can help understand the dynamics of cell walls to design root microbiomes to function in agroecosystems.
|
|
Scooped by
mhryu@live.com
Today, 1:42 AM
|
Against the backdrop of the growing global demand for green and sustainable biotechnology, efficiently and cost-effectively expressing enzyme proteins in multi-enzyme systems remains challenging. Drawing inspiration from the high-level expression of glucose-6-phosphate isomerase from E. coli in Bacillus subtilis, this study employed a region truncation analysis strategy based on three-dimensional structures to identify and develop a 30-amino acid N-terminal microtag, N30. When applied to enzyme expression in the artificial starch anabolic pathway (ASAP), the N30 tag enabled secretion levels of around 10 g/L for eight key enzymes. Significantly, N30(M22D)-TpiA reached 15 g/L in fed-batch fermentation, establishing an extremely efficient secretory expression system for ASAP enzymes. Furthermore, N30 was found to achieve detectable expression of multiple eukaryotic proteins that were previously unexpressible in Bacillus subtilis. Meanwhile, the fusion expression of rate-limiting enzymes in the synthetic pathway increased the titers of riboflavin and menaquinone-7 by 2.4- and 2.3-fold, respectively demonstrating the versatility and efficiency of N30 in protein synthesis and metabolic engineering applications. N30 provides a compact, activity-preserving, and scalable solution for the synthesis of enzymes for sustainable CO2 conversion, starch bioconversion, and other applications.
|
|
Scooped by
mhryu@live.com
Today, 1:37 AM
|
Antimicrobial resistance (AMR) poses a significant global health challenge, with the environment serving as a crucial reservoir and conduit for resistance determinants. Although antibiotic resistance genes (ARGs) have been extensively studied in environmental contexts, systematic approaches for assessing and prioritizing the risks associated with mobile genetic elements (MGEs), such as plasmids, phages, transposons, and integrative elements (IEs), remain unclear. To address this gap, we present MobiRes, an open-source computational framework designed to predict resistome risk by integrating information from the mobilome and microbiome. The pipeline was evaluated using a wide range of publicly available metagenomic datasets spanning diverse environments, including wastewater, poultry, soil, sediments, and human fecal samples. To validate the framework, statistical analyses and machine learning models were applied to evaluate the role of MGEs in driving ARG dissemination. The pipeline identified transposons as the dominant MGE class while capturing environment-specific variation in plasmid, phage, and IE -associated ARGs. Transposon-associated ARGs showed the most consistent environmental differentiation (ANOVA p = 0.0017; Kruskal–Wallis p = 0.018), whereas plasmid and phage-associated ARGs varied moderately ( p = 0.015–0.040) and IE-associated ARGs remained comparatively stable across environments ( p > 0.05). The Random Forest (RF) model achieved an AUC of 0.82, and subsequent feature importance and SHapley Additive exPlanations (SHAP) analyses revealed that transposon abundance is the primary factor driving ARG dissemination across diverse environments. By integrating host, mobility, and ecological factors, MobiRes provides a scalable and One Health–oriented framework for comprehensive AMR risk assessment. https://github.com/santhiyakc17/MobiRes_Pipeline.
|
|
Scooped by
mhryu@live.com
Today, 1:28 AM
|
Prokaryotic Argonaute (pAgo) proteins constitute an evolutionarily ancient nuclease family that is rapidly maturing into a versatile molecular toolkit rivaling CRISPR-Cas. This review synthesizes recent advances in pAgo biology and biotechnology, tracing their phylogeny across thermophilic, mesophilic, and psychrotolerant lineages and highlighting temperature-adapted catalytic signatures that diverge from eukaryotic Agos. In vivo studies reveal pAgo roles in gDNA-guided host defense, transcriptional silencing and recombination, all executed through programmable DNA- or RNA-guided nuclease activity. We detail how guide length, 5′ nucleotide identity, divalent cations and accessory factors modulate cleavage efficiency, enabling rational optimization. The review then maps the explosion of pAgo-based biosensing platforms, including selective nucleic acid enrichment platforms, ultrasensitive pathogen detection methods, programmable DNA cloning systems, and high-resolution imaging techniques. Their independence from protospacer adjacent motifs (PAMs), stable DNA guides, multi-turnover kinetics, and broad thermal tolerance position pAgos as ideal complements to CRISPR systems. Finally, we outline current limitations and future directions, including the discovery and engineering of novel variants, elucidation of guide-generation mechanisms, and development of next-generation gene-editing tools, aiming to accelerate translation of these versatile enzymes into practical biotechnological and therapeutic translation.
|
|
Scooped by
mhryu@live.com
Today, 1:18 AM
|
Unicellular microorganisms can make a transition to multicellular states that enhance survival under environmental fluctuations. In bacteria, one of these states is the biofilm, defined by the production of an extracellular matrix. Although biofilm maturation and dispersion have been extensively studied, where and how initial matrix production is induced within a growing population remains largely unknown. Here we show that production of colanic acid, an important matrix component, is initiated around topological defects, where cell orientation mismatches and growth-induced pressure builds up, in bacterial monolayers. Using E. coli reporting mechanically induced production of colanic acid in response to cell contact and deformation, we found matrix production accompanied by out-of-plane growth under agar-pad confinement. Controlling confinement geometry using microfluidic devices dictated the positions of topological defects and thereby localized regions of high matrix production. These findings reveal that the cell orientation patterning spatially organizes mechanical cues to induce matrix production for biofilm initiation of bacteria.
|
|
Scooped by
mhryu@live.com
Today, 1:05 AM
|
The scalability of phage therapy as a viable alternative or complement to antibiotics is limited by the labor-intensive experimental screening required to identify compatible phage-bacterium pairs. To accelerate this discovery process, we propose FoundedPBI, an ensemble deep learning approach that leverages the emergent capabilities of genomic foundation models, large language models pre-trained on vast DNA corpuses to predict phage-bacterium interactions from DNA sequences alone. We employ an ensemble strategy that aggregates outputs from three state-of-the-art DNA language models into a unified meta-embedding, which is then processed by a neural classifier. Our approach makes two key contributions: (1) We demonstrate that performing ensemble learning across models trained on different genomic data i.e., prokaryotic (Nucleotide Transformer v2, DNABERT-2) and bacteriophage (MegaDNA) genomes, captures partially-orthogonal biological signals, yielding 6% F1-score improvement over the best individual model. (2) We adapt long-context NLP aggregation strategies to handle whole bacterial and phage genomes (up to 5M base pairs) that exceed the foundation models context windows (12-96K bp) by a factor of 50-100, a critical challenge largely unaddressed in prior genomic deep learning work. On the PredPHI benchmark, FoundedPBI achieves a 76% F1-score outperforming the current state-of-the-art (PBIP) by 7%. On our internal dataset (CI4CB), we achieve 93% F1-score, improving our previous best methods by 4%. These results demonstrate that ensemble learning with proper long-context handling enables effective knowledge transfer of genomic foundation models to specialized prediction tasks.
|
|
Scooped by
mhryu@live.com
March 26, 11:47 PM
|
The canonical amino acid alphabet has remained remarkably stable since life’s early stages, yet the factors that shaped its emergence remain debated. Early views emphasized prebiotic availability and the expansion of metabolic pathways, but recent advances (particularly from protein biophysics and deep phylogenetics) have brought new perspectives to this question. Together, these views agree that the canonical alphabet emerged from a chemically restricted repertoire and was gradually tightened by metabolic innovation and selection for foldable, functional proteins. However, these approaches can yield inconsistent trajectories, underscoring how much remains unresolved. Here, we compare insights from four lines of evidence, highlight their limitations, and argue that our chronology of amino acid recruitment should be based on where the approaches converge.
|
|
Scooped by
mhryu@live.com
March 26, 11:09 PM
|
Bacterial surface polysaccharides are versatile structures that provide specificity to the behavior and interactions of a given bacterial strain. One surface polysaccharide displayed on the organism Bacteroides fragilis, Capsular Polysaccharide A, has been implicated as a potential therapeutic for autoimmune disorders. This polymer is composed of repeating units of the tetrasaccharide 2-acetamido-4-amino-2,4,6-trideoxygalactopyranose (AATGal), 4,6-O-pyruvate-galactopyranose (PyrGal), N-acetylgalactosamine (GalNAc), and galactofuranose (Galf). While this and other bacterial surface polysaccharides are attractive to study and apply to biomedicine, it can be difficult to acquire quick, inexpensive access to these pure materials. In this work, we developed a recombinant expression system in E. coli for the stepwise production of the CPSA polymer. A series of sequential plasmids were prepared, each incorporating successive genes required for CPSA biosynthesis. Using these iterative plasmids, we were able to observe production of the CPSA repeating unit and precursors by liquid chromatography mass spectrometry (LC-MS) analysis of cell lysates. We found that it was critical to include the CPSA polymerase but not the flippase, indicating that a native E. coli flippase could support polymer production. We also provide evidence that the CPSA polymer produced by E. coli can be ligated to LPS by the E. coli WaaL ligase, and deletion of this gene led to the formation of a water-soluble polymer. Overall, this work describes the first recombinant system for CPSA production and outlines a key strategy for the production of complex glycopolymers.
|
|
Scooped by
mhryu@live.com
March 26, 9:09 PM
|
The assembly of newly synthesized proteins into functionally active oligomers has long been regarded as a posttranslational process driven by random collision of subunits. However, growing evidence indicates that, for many proteins, assembly occurs cotranslationally, tightly coupling synthesis, folding, and subunit assembly. This fundamentally different mechanism enables the spatial and temporal coordination of assembly, promotes the hierarchical formation of multisubunit assemblies, enhances the stability of involved subunits, enlarges the space of feasible protein structures including complexes with intertwined subunits, and has profound effects on protein evolution and function. In this review, we describe the molecular mechanisms, cellular requirements, and functional implications of cotranslational assembly and discuss its relevance to human disease, its evolutionary significance, and its transformative potential in synthetic biology and recombinant protein production.
|
|
Scooped by
mhryu@live.com
March 26, 8:46 PM
|
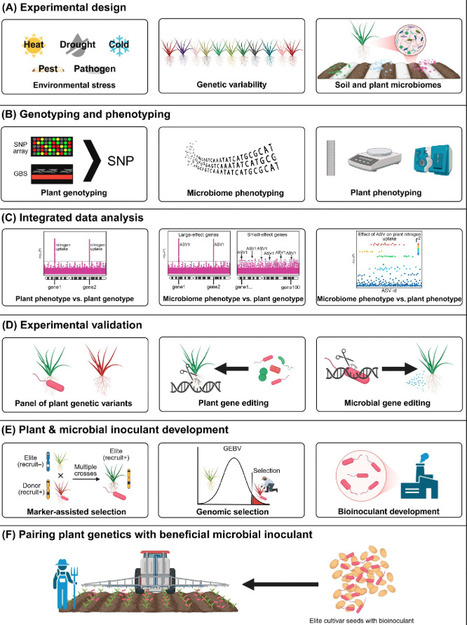
Plants constantly form associations with microorganisms, and some are vital for plant performance, especially under stress conditions. Although some microorganisms have been developed into commercial bioinoculant products, their associations with plants can be transient, and their efficacy is often inconsistent in the field. Here, we propose a framework and key research steps needed for plant breeders and microbiologists to work together to match plant genetics with compatible microbial inoculants. Recent studies have successfully identified plant genetic factors that play a role in the recruitment of beneficial microorganisms, and many associated technologies are ready for implementation towards this goal. This innovative and collaborative approach could provide novel, enduring plant–microbiome associations for environmental sustainability and food security under a changing climate.
|
|
Scooped by
mhryu@live.com
March 26, 8:15 PM
|
Antimicrobial agents play a vital role in human and environmental health, with applications spanning medicine, food preservation, agriculture, and biotechnology. Among them, enzybiotics enzyme-based antimicrobials have emerged as powerful alternatives to conventional antibiotics due to their targeted mechanisms and lower propensity for resistance. Beyond their medical relevance, enzybiotics have emerging applications in food preservation, animal health, and agriculture, thereby broadening their industrial and environmental value. To support the discovery and characterization of these versatile biomolecules, we present the first genome-resolved metagenomic gene and protein targeted enzybiotic catalog focused on enzybiotics, derived from diverse environmental microbiomes. The Microbial Enzybiotic Gene and Protein Catalog (MiGPC), integrates 15 whole-metagenome datasets from oceans, soils, fecal samples, vegetation, and plastic-contaminated environments, capturing a wide ecological spectrum. Enzybiotic sequences were compiled through a hybrid strategy combining public database mining and manual literature curation, yielding over 136,000 enzybiotic sequences, 7654 metagenome-assembled genomes (MAGs), and ~ 100 million unique genes and proteins. MiGPC integrates taxonomic and enzybiotic gene profiles, offering a robust platform for the discovery, annotation, and ecological mapping of antimicrobial enzymes. Functional analyses using KEGG and eggNOG revealed that approximately 62% of the genes remained uncharacterized, highlighting a rich source of potentially novel functions. Glycoside hydrolases and glycosyl transferases were the most prevalent CAZyme families, while the dominant enzybiotic-producing taxa belonged primarily to the Pseudomonadota and Bacillota phyla. Statistical modeling uncovered two major ecological clusters that distinguished polluted from relatively pristine environments. MiGPC enables high-throughput screening of previously unexplored metagenomes, facilitating the identification of novel antimicrobial agents from under characterized ecosystems. Overall, MiGPC represents a landmark resource that will support multi-omics research, microbial ecology, and the development of next-generation biotechnological solutions based on enzybiotics.
|
|
Scooped by
mhryu@live.com
March 26, 7:40 PM
|
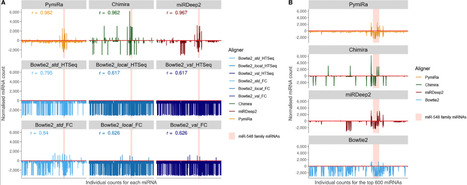
Small non-coding RNAs (sncRNA; < 200 nucleotide length) are of increasing research interest due to their key regulatory roles in a host of fundamental biological processes. For example, microRNAs (miRNAs), a specific class of sncRNAs, regulate gene expression through messenger RNA interactions, and their dysregulation is associated with disease. Classifying sncRNAs is an important bioinformatic task in small RNA-sequencing pipelines. Here we have developed an aligner called PymiRa, written in Python, to identify and quantify miRNAs from FASTA/FASTQ sequencing files. Unlike other approaches, PymiRa utilizes a Burrows-Wheeler algorithm to align an input file against a reference hairpin precursor FASTA file derived from miRBase, the online miRNA registry, permitting up to two mismatches at the 3’ end of a read. Previous tools used either a Burrows-Wheeler genome alignment or dynamic programming alignment to precursors; we demonstrate that combining both approaches yields improved results and efficiency. Importantly, the PymiRa aligner accounts for 3’ post-transcriptional modifications to miRNAs that typically occur. PymiRa is a fast, accurate, and publicly accessible aligner available via GitHub and/or a webserver for sncRNA identification, including miRNAs, enabling accurate counts to be produced as part of a small RNA-sequencing pipeline. PymiRa will undergo relevant revisions over time e.g., with miRBase version updates. The PymiRa aligner will facilitate a deeper biological understanding of the landscape of sncRNA expression in normal physiological conditions and their dysregulation in disease states, including cancer.
|
|
Scooped by
mhryu@live.com
March 26, 7:09 PM
|
The ability to precisely control gene expression is fundamental to studying biological processes. Using site-specific recombinases such as FLP, gene expression can be controlled, albeit with limited spatio-temporal precision. We develop a photocaged FLP recombinase, which can be precisely controlled using light, and we demonstrate its efficacy in Caenorhabditis elegans. We use genetic code expansion to incorporate photocaged amino acids into FLP, replacing critical residues in the active site with their photocaged counterparts. Photocaged FLP displays no detectable background activity, and brief illumination can be used to activate FLP with near 100% efficiency. We show that photocaged FLP can be activated by light between 365 and 435 nm, and that it is not activated by light above 450 nm, making it fully compatible with wavelengths commonly used for imaging and optogenetics. Furthermore, we demonstrate that photocaged FLP can be used to switch on expression of target genes in individual cells within the animal using a standard 405 nm microscope-mounted laser to deliver the activating light. Activation by laser requires illumination times of <10 ms per cell. Thus, we have developed a straightforward and efficient tool to precisely control gene expression in the multicellular organism C. elegans.
|
|
|
Scooped by
mhryu@live.com
Today, 1:47 AM
|
Recent advances in peptide inhibitors have highlighted their therapeutic potential across cancers, neurodegenerative, cardiovascular, and metabolic diseases, driving the need for efficient molecular genetic methods to generate large peptide libraries. This study introduces improvements to existing protocols to enhance product yield, expand library coverage, and reduce time and cost. The approach incorporates Single Self-Annealing PCR Primers (SSAP) to replace traditional primer pairs, alongside optimized PCR conditions to boost specificity and eliminate non-specific fragments. Additional refinements in cloning and transformation steps significantly increased the efficiency of E. coli DH5α and 10-beta strains, outperforming commercial high-efficiency competent cells. SSAP produced clean, desired PCR products that may improve, theoretically, ligation efficiency by 50% and increased transformation output twelve-fold, resulting in a purer and more accurate library. Overall, these methodological enhancements provide a more robust, cost-effective strategy for constructing large peptide libraries and offer versatile tools applicable to similar molecular genetics experiments.
|
|
Scooped by
mhryu@live.com
Today, 1:40 AM
|
Trichodermin, a sesquiterpenoid with notable antifungal, plant growth-regulating, and antitumor activities, holds broad application potential in medicine and agriculture. However, its low native production in Trichoderma presents a significant bottleneck, hindering the large-scale utilization and development of high-value pharmaceutical derivatives. In this study, a systematic metabolic engineering framework was established in the native producer T. taxi to enhance trichodermin production. A foundational genetic toolbox was first developed, enabling the robust overexpression, knockout, and knockdown of genes. This system was validated using the tri5 gene as a model, facilitating iterative rounds of strain engineering. A subsequent key outcome was the identification of the ABC transporter G5728 as a central trichodermin efflux pump, achieved through an integrated in silico strategy combining homology mining, transcriptomics, and molecular docking. Overexpression of G5728 resulted in a 70.9% increase in trichodermin production, achieving a titer of 2.0 g/L. Building on this, four key modules were systematically engineered: enhancement of acetyl-CoA precursor supply, MVA pathway flux, and trichodermin biosynthesis, along with the downregulation of competing pathways. This integrated approach led to the construction of the high-yield T. taxi strain LH58, which achieved a trichodermin titer of 7.22 g/L in a 5-L bioreactor. This represents a substantial advancement in productivity, highlighting its potential for industrial-scale biomanufacturing. This work provides a reliable platform for the sustainable production of trichodermin and offers a transferable strategy for optimizing terpenoid biosynthesis in microbial hosts.
|
|
Scooped by
mhryu@live.com
Today, 1:35 AM
|
Chaperone/Usher (CU) pathway pili constitute the most diverse class of pili in Gram-negative bacteria. Each pilus filament is assembled from thousands of pilin subunits, and thousands of such filaments can be simultaneously displayed on the bacterial surface, where they mediate essential functions related to environmental adaptation. Owing to their distinctive architectural features, including micrometer-scale length, high copy number, and repetitive subunit organization, CU pili possess intrinsic potential as platforms for bacterial surface display in biotechnology and synthetic biology. Early efforts in the 1980s and 1990s explored CU pili as display scaffolds but failed to yield broadly applicable or robust systems, largely due to limited understanding of pilus assembly mechanisms and the absence of high-resolution structural information. In recent years, the rapid accumulation of atomic and near-atomic resolution structures of CU pilins and their assembly intermediates, together with advances in protein structure prediction, has fundamentally reshaped the landscape of pilus engineering. In this review, we use structurally characterized CU pili from E. coli, Salmonella and Yersinia pestis as reference models to systematically analyze the structural constraints governing CU pilus-based display. We reinterpret prior display strategies through a unified structural framework, distill conserved design principles that define engineering permissiveness, and propose practical engineering approaches for expanding CU pili into versatile and tunable surface display platforms. Collectively, this work provides a structure-guided framework for assessing feasibility and guiding the rational engineering of CU pilus display systems within the synthetic biology toolbox.
|
|
Scooped by
mhryu@live.com
Today, 1:20 AM
|
Transformer-based genomic sequence models represent an emerging frontier in computational biology. Yet, their embeddings have not yet shown the same level of predictive power as natural and protein language models, indicating a gap between current implementations and theoretical promise. Existing benchmarks for DNA language models primarily focus on classifying regulatory elements in eukaryotic genomes, leaving open the fundamental question of whether these models learn sequence-level features across whole genomes. We introduce LAMBDA, a benchmark designed to rigorously evaluate genome language model embeddings through phage-bacteria sequence discrimination across four categories of increasing complexity: probing tasks, fine-tuning assessments, diagnostic tests, and genome-wide prophage detection. Our comprehensive analysis of current genomic language models provides novel insights into the importance of training data quality relative to model size, the need for domain-specific training, and the application of genomic language models for detecting prophage sequences. This benchmark represents a challenging genomic annotation task in the bacterial domain and addresses a key computational problem with direct relevance to microbiology and medicine.
|
|
Scooped by
mhryu@live.com
Today, 1:14 AM
|
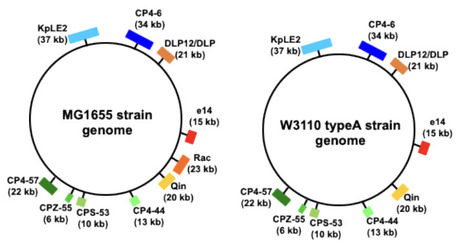
Bacteria have evolved sophisticated mechanisms to sense and adapt to environmental shifts, particularly when transitioning into the stable, warm temperatures of a host organism. E. coli, a primary inhabitant of the warm-blooded host gut, must rapidly initiate growth upon entry to ensure reproductive success. In this study, we demonstrate that the specific growth rate at the onset of the logarithmic phase (defined as μ at ODratio=0.2) is specifically stimulated across a medium temperature range (30°C–42°C), peaking near the physiological temperature of 37°C. This adaptive response is strain-specific and depends on both the histone-like nucleoid-associated protein (NAP) H-NS and the presence of the Rac prophage. Using high-frequency automated growth monitoring and statistical modeling, we redefine H-NS not merely as a gene silencer but as a critical "nucleoid structural organizer". Our results indicate that H-NS undergoes a conformational switch at approximately 37°C, transitioning into a parallel form that provides the necessary physical scaffold for nucleoid reorganization. This reorganization is essential for coordinating transcription and replication during the rapid onset of growth. Crucially, we resolve the "silencing paradox": while H-NS silencing is traditionally thought to be weakened at 37°C, hns-deficient mutants grow significantly slower because they lack this essential structural scaffold. We conclude that the H-NS-mediated physical organization of the genome is more critical for host adaptation than the mere de-repression of the genomic reservoir, enabling E. coli to effectively transition into a high-growth state for successful host colonization.
|
|
Scooped by
mhryu@live.com
Today, 12:29 AM
|
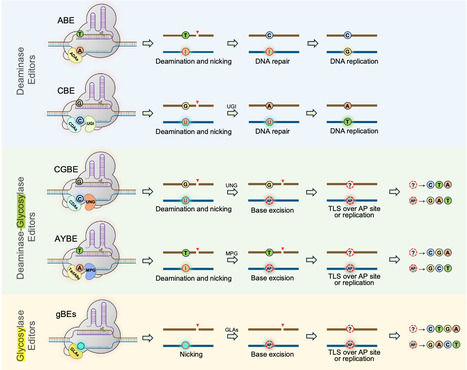
Base editors introduce defined DNA lesions rather than directly rewriting nucleotides, and these lesions must be processed by cellular DNA repair pathways. In glycosylase-mediated base editing, apurinic/apyrimidinic sites (AP sites) represent a critical decision point where the choice of repair pathway determines whether the outcome is a desired substitution or an unintended byproduct. Although product heterogeneity has long been considered an intrinsic limitation of base editing, recent mechanistic studies suggest competition among repair polymerases represents a major contributor. Evidence indicates translesion synthesis polymerases can serve as tunable resolvers by preferentially inserting specific nucleotides opposite AP sites. Engineering their recruitment and insertion preferences may shift base editing from a stochastic repair-dependent process toward a more predictable mode of nucleotide incorporation.
|
|
Scooped by
mhryu@live.com
March 26, 11:36 PM
|
Protein expression is an important aspect of synthetic biology, where host selection critically impacts the production efficiency. V. natriegens has emerged as a promising prokaryotic host due to its exceptionally rapid growth rate and broad substrate metabolic spectrum. Benefiting from the advances in annotation on microbial genomes and metabolic pathways, as well as the development of molecular biology tools, continuous progress has been made in bioproduction using V. natriegens. In this Perspective, we provide a systematic introduction of the basic biological properties of V. natriegens and present the engineered V. natriegens chassis which are currently used for protein expression. We summarize the latest synthetic biology components and genetic tools specifically designed for the modification of V. natriegens. Especially, we focus on the most recent study and application achievements of V. natriegens in in vivo and in vitro protein expression, which has received limited attention in previous reviews. This review aims to present researchers with a comprehensive overview of using V. natriegens for protein expression, highlight its advantages and application prospects, and present some challenges and future perspectives in this field, so as to promote wider research in the synthetic biology field.
|
|
Scooped by
mhryu@live.com
March 26, 9:17 PM
|
mRNA vaccines benefit from rapid design, scalable manufacturing and strong safety profiles, and they are being preclinically and clinically explored for various infectious diseases, cancer and autoimmune disorders. However, global access to mRNA vaccines remains limited by technical, logistical, economic, regulatory and ethical challenges. In this Review, we discuss the current mRNA vaccine landscape, from approved products to emerging candidates, and outline key barriers to equitable access. We highlight engineering strategies to address these barriers, including thermostability improvements, alternative administration routes, new delivery systems and alternative RNA platforms, such as self-amplifying and circular RNA, as well as the integration of artificial intelligence- and machine learning-based design and optimization. We also discuss the regulatory, ethical and social dimensions of vaccine deployment, as well as the importance of building public trust and community engagement. Coordinated technical and policy advances are essential to remove barriers, enhance access and advance global health equity. mRNA vaccines combine rapid design, scalable manufacturing and strong safety profiles with applications across infectious diseases, cancer and autoimmune disorders. However, global access to mRNA vaccines remains limited. This Review explores how achieving equitable access depends on overcoming technical, logistical, economic and regulatory barriers through innovations in thermostability, delivery systems, alternative RNA platforms, artificial intelligence-driven design and optimization, and coordinated policy strategies.
|
|
Scooped by
mhryu@live.com
March 26, 9:06 PM
|
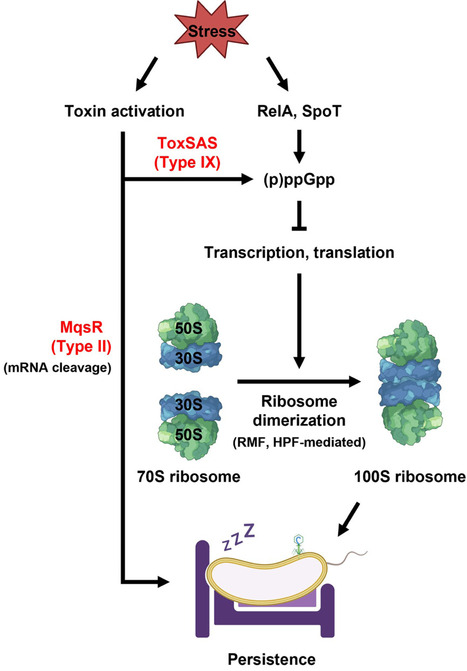
The toxin–antitoxin (TA) field continues to advance rapidly, especially given the recognition of TA systems as one of the most prevalent phage defenses. These systems are omnipresent in both bacteria and archaea and are currently classified into eight types. Here, we propose an additional type IX class, in which either the toxin or antitoxin utilizes nucleotide-based signaling. By establishing the new type IX TA system, the framework for the organization of TA systems, which is based on the diverse biochemical functions of both the toxins and antitoxins across the nine types, will be refined and aid in their discovery and understanding. In addition, we highlight the increasing evidence that TA systems are related to persistence and are activated by conformational changes in protein complexes.
|
|
Scooped by
mhryu@live.com
March 26, 8:43 PM
|
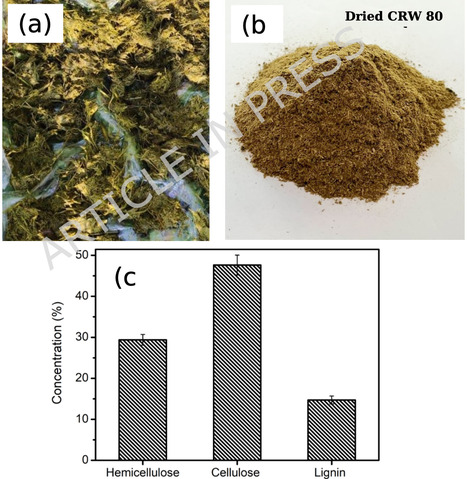
Cow rumen waste (CRW), a slaughterhouse by-product rich in lignocellulose, can be directly converted into bioethanol without harsh chemical pretreatment or additional microbial inocula. In this study, we developed an integrated enzymatic hydrolysis and fermentation process to produce bioethanol from CRW using Viscozyme Cassava CL and Saccharomyces cerevisiae. Key process parameters for enzymatic hydrolysis (enzyme concentration, pH, and substrate loading) were optimized, with 3 wt% enzyme at pH 5.0 and 50 g L− 1 CRW yielding the highest glucose release. The enzymatic hydrolysate was then fermented by S. cerevisiae under optimal conditions (inoculum 3 wt%, 30–35 °C, pH 5.0), producing an ethanol concentration of 4.79 g L− 1 with negligible residual sugar. These results demonstrate the feasibility of one-pot conversion of CRW to bioethanol. The novel enzyme-assisted process is scalable and low-cost, offering a sustainable strategy to valorize slaughterhouse waste into renewable bioethanol.
|
|
Scooped by
mhryu@live.com
March 26, 8:06 PM
|
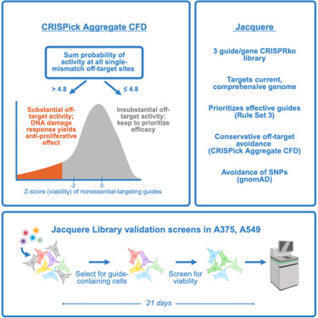
The continued development of high-dimensional CRISPR screen readouts, such as single-cell RNA sequencing and high-content imaging, necessitates compact libraries to enable functional interrogation at genome scale. Improved genome annotations cause library deprecation over time, further motivating an updated genome-wide design effort. Additionally, while on-target efficacy and off-target avoidance are often optimized in isolation, we lack a robust framework for simultaneously weighing and balancing these competing priorities. Here, we present a selection strategy that identifies guides with sufficient off-target activity to justify omission from the library, thus avoiding the unnecessary exclusion of active guides, allowing the inclusion of those with maximal on-target activity. We create, validate, and make available to the community the Jacquere library for knockout screens of the human genome, as well as its mouse counterpart, Julianna, to facilitate gene function discovery at scale.
|
|
Scooped by
mhryu@live.com
March 26, 7:35 PM
|
Current one-pot CRISPR diagnostics are limited by the cis-cleavage activity of Cas nucleases, which leads to amplicon degradation during amplification. Here, we report a streamlined strategy that overcomes this limitation. By integrating a bipartite split-crRNA into Cas12a (SCas12a), we separate target recognition from PAM dependency and completely eliminate cis-cleavage while preserving robust trans-cleavage. This strategy is broadly applicable for one-pot testing, compatible with recombinase polymerase amplification, RT–RPA, and loop-mediated isothermal amplification LAMP, as well as multiple Cas12a orthologs, including As, Lb, and Ct Cas12a. Moreover, the SCas12a accelerates one-pot testing with 100–1000-fold improved sensitivity and achieves >10-fold reduction in time-to-signal, enabling detection of targets at attomolar levels within 30 min. Additionally, it provides single-base resolution with up to 91-fold selectivity. The system has been successfully applied to detect HPV16, SARS-CoV-2, and TP53 SNPs in clinical samples. Together, we have developed a PAM-independent and cis-cleavage-free one-pot Cas12a assay, which holds strong potential for point-of-care diagnostics.
|























genetic part