 Your new post is loading...

|
Scooped by
mhryu@live.com
Today, 12:55 PM
|
Although it is increasingly recognized that anthropogenic chemicals modulate horizontal gene transfer (HGT), the nature of these interactions is often more complex than a simple promotion or inhibition. The potential for a single chemical to exert opposing, concentration-dependent effects represent a critical and less-explored frontier in microbial ecology. Here, we investigate the last-resort antibiotic polymyxin B, a membrane-targeting peptide, and reveal a concentration-dependent, biphasic regulation of plasmid conjugation. Sub-inhibitory concentrations (0.125-0.5 mg/L) consistently inhibited the transfer of antibiotic resistance genes (ARGs) by up to 65.4%, whereas bactericidal concentrations (≥ 1 mg/L) strongly promoted it by up to 15.9-fold. This regulatory switch is driven by distinct physiological states: low-level exposure triggers defensive responses including reduced membrane permeability, whereas high-level exposure causes catastrophic membrane damage, inducing a synergistic stress response involving oxidative damage (>2-fold ROS increase) and a surge in cellular energy (up to 83.0% ATP increase) that facilitates HGT. High-concentration polymyxin B also promotes plasmid transfer in complex microbial communities derived from activated-sludge biofilms. Our findings reveal a new paradigm for the interaction between chemical stressors and microbial evolution, demonstrating that the ecological impact of contaminants on HGT cannot be predicted by monotonic models and highlighting the role of environmental hotspots in shaping the dissemination of antibiotic resistome.
|
|
Scooped by
mhryu@live.com
Today, 12:47 PM
|
Terpenoids are highly abundant and valuable natural products, yet high-throughput screening platforms for microbial producers remain scarce. To address this, we developed a microfluidic screening platform leveraging the intrinsic fluorescence of β-carotene. Using an engineered yeast chassis (YsL4) with a reinforced mevalonate pathway, we optimized cultivation conditions and achieved perfect (100%) phenotypic specificity in a 1:1 coencapsulation assay between producer (YsL4) and nonproducer (Ys011) strains. Screening a genome-wide CRISPRa/i library via multiround fluorescence-activated droplet sorting identified 15 target genes. Validation confirmed that 10 targets (5 for overexpression, 10 for knockout) significantly enhanced β-carotene production. A key mutant, YL19 (ΔHMO1), achieved a titer of 33.71 mg/L in shake-flasks─a 149.15% increase over the parent strain─with a concurrent lycopene reduction indicating redirected carbon flux toward β-carotene biosynthesis. This integrated platform enhances screening efficiency and provides a new paradigm for identifying critical terpenoid biosynthetic targets.
|
|
Scooped by
mhryu@live.com
Today, 12:27 PM
|
Characterizing CRISPR interference (CRISPRi) phenotypes presents a fundamental temporal challenge: pre–existing overabundance of target proteins can mask early silencing, requiring extended growth for dilution, yet prolonged repression rapidly selects for escaper mutants. To resolve this, we integrated a tightly regulated CRISPRi system in Pseudomonas putida with an automated mini-bioreactor platform operating in turbidostat mode. By maintaining continuous exponential growth, we mapped the exact temporal dynamics of essential gene silencing. We identified a critical observation window between 17 and 27 hours (7–9.5 cell doublings) where repression exerts its maximum physiological impact, directly preceding population takeover by target-site mutated escapers. Applying this workflow to the arginine biosynthesis pathway, multi-omics profiling disentangled transient physiological buffering from long-term mutational events, and revealing that argH and argG knockdowns trigger highly diverse metabolomic perturbations. This scalable framework overcomes batch culture limitations, ensuring precise temporal control for accurate phenotypic characterization and reliable functional genomics.
|
|
Scooped by
mhryu@live.com
Today, 12:18 PM
|
This study presents dAMN, a hybrid neural-mechanistic model that integrates neural networks with genome-scale dynamic flux balance analysis (dFBA) to predict bacterial growth curves across diverse nutrient environments. dAMN uses neural networks to infer dynamic behavior from initial metabolite concentrations, while mechanistic constraints ensure stoichiometric and thermodynamic consistency based on genome scale metabolic models. dAMN is trained on E. coli and P. putida experimental growth data from media containing various combinations of sugars, amino acids, and nucleobases, and evaluated on two test sets: one for forecasting over time and another for predicting growth dynamics on unseen media. dAMN achieved high predictive power (R > 0.9), successfully reproducing growth and substrate depletion dynamics including acetate overflow and glucose-acetate consumption shift for E. coli. An interesting innovation of dAMN is the treatment of the lag phase, enabling realistic adaptation dynamics absent from standard dFBA models. dAMN stands out for its ability to generalize across combinatorial nutrient inputs and produce full growth-curve predictions from minimal input data.
|
|
Scooped by
mhryu@live.com
Today, 12:09 PM
|
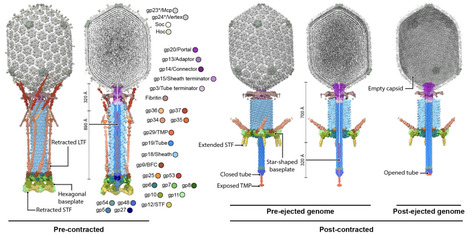
Successful viral infection requires efficient adsorption to the target cell, followed by membrane penetration for genome translocation into the host cytoplasm. Bacteriophage T4 initiates infection by recognising E. coli surface receptors via its long tail fibres. Receptor binding triggers sequential conformational changes that culminate in tail sheath contraction and genome delivery through viral channels spanning the host cell envelope. Despite extensive structural studies, the mechanism of genome translocation by tailed phages remains unclear. Here, using cryo-electron microscopy, we resolved structures of bacteriophage T4 at discrete stages. This revealed how long tail fibre extension affects the baseplate to initiate tail contraction, and the domain architecture of the tape measure protein and its involvement in channel formation and genome translocation. Furthermore, we demonstrate that the virus-encoded superinfection exclusion protein Imm binds to the tape measure protein to prevent secondary infections. Our findings reveal the mechanism of tape measure protein-mediated membrane penetration, offering insights into the coordinated process of genome delivery and phage-encoded superinfection exclusion proteins that prevent genome translocation.
|
|
Scooped by
mhryu@live.com
Today, 11:57 AM
|
RNA interference (RNAi) shows great potential to protect agricultural crops against a variety of fungal diseases. However, protection efficiencies reported by empirical RNAi studies can vary greatly, and our understanding of the factors responsible for this variance remains limited. In this meta-analysis, we evaluated 89 studies that compare the efficiency of host-induced gene silencing (HIGS) and spray-induced gene silencing (SIGS) in controlling fungal diseases, focusing on biotrophic, hemibiotrophic, and necrotrophic fungi, the use of formulations and the dsRNA design as explanatory factors for differences between reported efficiency values. Our results indicate that SIGS is slightly more effective, particularly in biotrophs. Somewhat surprisingly, we did not find significant efficiency differences between SIGS studies that used formulations and those that applied naked dsRNA. We also considered the effect of various parameters describing RNA design. Differences in dsRNA length and the number of constructs, and number of targets showed no consistent significant effect on resistance in either HIGS or SIGS. Interestingly, however, HIGS studies reported significantly higher efficiency when targeting genes closer to the 3` end and SIGS when targeting genes closer to the 5` end. We discuss reasons for the reported patterns, such as variability in dsRNA uptake mechanisms, intercellular trafficking and Dicer processing, and conclude that more research is needed to understand the biological mechanisms determining RNAi efficiency for fungal control.
|
|
Scooped by
mhryu@live.com
Today, 11:45 AM
|
Wood, once regarded primarily as a structural material, possesses rich physicochemical complexity that has long been underexplored. In the context of industrialization and carbon imbalance, it is now emerging as a renewable and multifunctional platform for green nano-technologies. Recent advances in wood nanotechnology have enabled the transformation of natural wood into programmable substrates with tailored nanoarchitectures, establishing it as a representative class of bio-based nanomaterials. This review systematically categorizes wood-specific nanoengineering strategies—including thermal carbonization, laser-induced graphenization, targeted delignification, nanomaterial integration, and mechanical processing—highlighting their mechanisms and impacts on wood’s multiscale structural and functional properties. Importantly, these functionalization strategies can be flexibly combined in a modular, “Lego-like” manner, enabling wood to be reconfigured and optimized for diverse application scenarios. We summarize recent progress in applying functionalized wood to sustainable technologies such as energy storage (e.g., metal-ion batteries, Zn–air systems, supercapacitors), water treatment (e.g., adsorption, photothermal filtration, catalytic degradation), and energy conversion (e.g., solar evaporation, ionic thermoelectrics, hydrovoltaics, and triboelectric nanogenerators). These studies reveal how nanoengineered wood structures can enable efficient charge transport, selective adsorption, and enhanced light-to-heat conversion. Finally, the review discusses current challenges—such as scalable fabrication, material integration, and long-term environmental stability—and outlines future directions for the development of wood-based platforms in next-generation green energy and environmental systems.
|
|
Scooped by
mhryu@live.com
Today, 11:32 AM
|
Polyamines are ancient metabolites that serve critical functions in maintaining epithelial integrity, regulating immune response, and supporting healthy aging. The gut microbiota actively synthesizes and converts polyamines, while host factors such as inflammation, barrier function, and nutritional status dynamically modulate this metabolic network. Disruption of this host–microbiota axis reduces polyamine availability, impairs barrier function, and exacerbates inflammation. In contrast, polyamines exert protective effects by promoting epithelial repair, modulating macrophage and T-cell responses, and enhancing autophagy-mediated tissue renewal and longevity. Recent advances in engineered probiotics, microbial small RNAs, and postbiotics further highlight the therapeutic potential of precisely modulating polyamine metabolism in clinical contexts such as inflammatory bowel disease, metabolic syndrome, and neurodegenerative conditions associated with aging.
|
|
Scooped by
mhryu@live.com
March 6, 5:53 PM
|
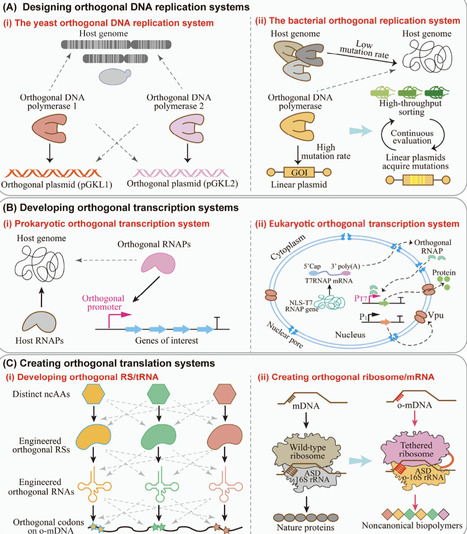
Microbial synthetic biology focuses on the application of rational engineering strategies to reprogram microbial cells, thereby providing them with novel functions to meet different requirements. However, this engineering process inherently disrupts natural metabolism, leading to increased complexity and unpredictability within the metabolic system. To address these challenges, a series of orthogonal strategies has been developed and implemented in the construction of orthogonal genetic systems, metabolic pathways, energy systems, and regulatory systems. This review summarizes recent advances and applications of orthogonal strategies in microbial synthetic biology. Finally, future research directions in orthogonal microbial synthetic biology are explored, aiming to provide new insights for future studies.
|
|
Scooped by
mhryu@live.com
March 6, 5:02 PM
|
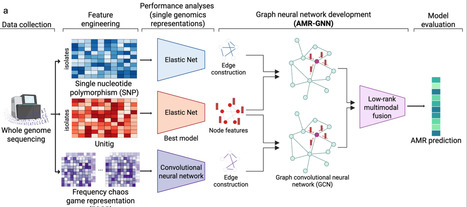
Whole-genome sequencing (WGS) data are an invaluable resource for understanding antimicrobial resistance (AMR) mechanisms. However, WGS data are high-dimensional and the lack of standardized genomic representations is a key barrier to AMR phenotype prediction. To fully explore these high-resolution data, we propose AMR-GNN, a graph deep learning-based framework that integrates multiple genomic representations with graph neural networks (GNN) to enable AMR phenotype prediction from genomic sequence data. We test AMR-GNN with Pseudomonas aeruginosa, a clinically relevant Gram-negative bacterial pathogen known for its complex AMR mechanisms. We present AMR-GNN as a proof-of-concept framework designed to address several key problems in AMR phenotype prediction with data-driven machine learning (ML) approaches, including using multiple genomic representations to enhance performance, to mitigate the influence of clonal relationships and to identify informative biomarkers to provide explainability. Follow-up validation on the largest publicly available dataset spanning both Gram-negative and Gram-positive pathogens highlights AMR-GNN’s broad applicability in detecting AMR in diverse and clinically relevant pathogen-drug combinations. Predicting antimicrobial resistance from bacterial genomic data is challenging. Here, the authors introduce AMR-GNN, a graph neural network that integrates multiple genomic representations to improve prediction, reduce clonal bias, and identify biomarkers.
|
|
Scooped by
mhryu@live.com
March 6, 1:59 PM
|
Evolving dynamic, multi-state, and computational protein functionalities is challenging because it requires selection pressure on all the states of a protein of interest (POI) and the transitions between them. To create a continuous directed evolution paradigm for such properties, we genetically engineered budding yeast for optogenetic input to switch a POI “on” and “off,” which, in turn, controls a Cdk1 cyclin that is essential for one cell-cycle stage but detrimental for another. The method, “optovolution,” generates dynamic selection pressure on POI cycling at the timescale of tens of minutes. We used it to evolve 19 new variants of the LOV transcription factor El222, including in vivo green-light-responsive variants allowing LOV color-multiplexing. Evolving the PhyB-Pif3 optogenetic system, we discovered that loss of YOR1 makes supplementing phycocyanobilin (PCB) unnecessary. Finally, we demonstrated the generality of the method by evolving a non-light-responsive AND gate (PEST-rtTA). Optovolution makes difficult-to-engineer protein functionalities continuously evolvable.
|
|
Scooped by
mhryu@live.com
March 6, 1:24 PM
|
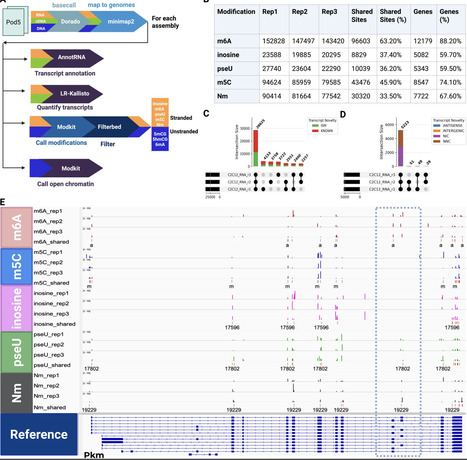
Oxford Nanopore (ONT) sequencing allows for the direct detection of RNA and DNA modifications from unamplified nucleic acids, which is a significant advantage over other platforms. However, the rapid updates to ONT basecalling models and the evolving landscape of computational tools for modification detection bring about challenges for reproducible and standardized analyses. To address these challenges, we developed Dogme to automate basecalling, alignment, modification detection, and transcript quantification. Dogme automates the reprocessing of ONT POD5 files by integrating basecalling using Dorado, read mapping using minimap2 and subsequent analysis steps such as running modkit. The pipeline supports three major types of sequencing data—direct RNA (dRNA), complementary DNA (cDNA), and genomic DNA (gDNA). Dogme facilitates detection of diverse RNA modifications supported by Dorado such as N6-methyladenosine (m6A), 5-methylcytosine (m5C), inosine, pseudouridine, 2’-O-methylation (Nm) and DNA methylation, while concurrently quantifying full-length transcript isoforms LR-Kallisto for transcript quantification for dRNA and cDNA. We applied Dogme to three separate mouse C2C12 myoblast replicates using direct RNA sequencing on MinION flow cells. We detected 96 603 m6A, 43 476 m5C, 8829 inosine, 10 055 pseudouridine, and 30 320 Nm sites in three biological replicates. The pipeline produced reproducible modification profiles and transcript expression levels across replicates, demonstrating its utility for integrative long-read transcriptomic and epigenomic analyses.
|
|
Scooped by
mhryu@live.com
March 6, 2:00 AM
|
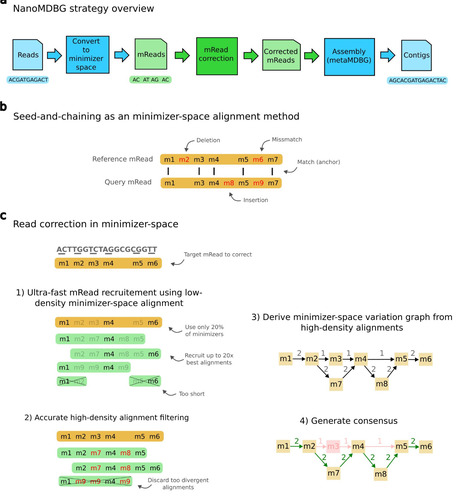
Third-generation long-read sequencing technologies, significantly improve metagenome assemblies. Highly accurate PacBio HiFi reads can yield hundreds of near-complete metagenome-assembled genomes (MAGs) from a single sample. Recently, the accuracy of the more cost-effective Oxford Nanopore Technologies (ONT) platform has increased to a per-base error rate of 1-2%. However, current metagenome assemblers are optimized for HiFi and do not scale to the large data sets that ONT enables. We present nanoMDBG, an evolution of metaMDBG, which supports the latest ONT reads through an error correction pre-processing step in minimizer-space. Across a range of ONT datasets, including a large 400 Gbp soil sample, nanoMDBG reconstructs up to twice as many high-quality MAGs as the next best ONT assembler, metaFlye, while requiring a third of the CPU time and memory. Critically, the latest ONT technology can now produce comparable MAG construction results as those obtained using PacBio HiFi at the same sequencing depth. Benoit et al. present nanoMDBG, a scalable metagenome assembler for the latest ONT long reads, developed by adding error correction to metaMDBG. They demonstrate superior performance to the state-of-the-art and equivalent results to PacBio HiFi reads.
|
|
|
Scooped by
mhryu@live.com
Today, 12:51 PM
|
For much of the 20th century, enzyme kinetic analysis relied on deriving simplified rate equations under the steady-state approximation and later by analytical integration of differential equations for transient kinetics. This approach has since been surpassed by computational methods using numerical integration of rate equations to directly fit experimental data based on a complete user-defined model. This paradigm shift removes the constraints imposed by solving analytical equations, enabling far greater flexibility in experimental design and model complexity. Modern global fitting methods allow data from diverse experiments to be analyzed simultaneously using the minimum number of parameters supported by the information content of the data set. Global data fitting is more than just an algorithm for data analysis─it represents a fundamental change in how we design and interpret experiments, and eliminates many of the restrictions, approximations, and ambiguities inherent to equation-based analyses. In this review, we describe the principles and practice of global data fitting, compare the outcomes to conventional equation-based methods, and demonstrate its power through examples involving multiple experiments with distinct conditions and readouts. We explain why the common practice of making measurements in triplicate introduces uncertainty and we outline advanced methods for rigorously estimating errors in measurement and for establishing robust confidence limits on fitted parameters.
|
|
Scooped by
mhryu@live.com
Today, 12:36 PM
|
Intermicrobial and host–microbial interactions are critical for the functioning of the gut microbiome, but few tools are available to measure these interactions in situ. Here we report a method for broad spatial sampling of microbiome–host interactions in the gut at high resolution (1 µm). This method combines enzymatic in situ polyadenylation of both bacterial and host RNA with spatial RNA sequencing to increase bacterial RNA recovery and enable transcriptomic analysis of low-abundance and spatially restricted microbial taxa. We benchmark the method against existing spatial transcriptomic workflows, demonstrating improved sensitivity and resolution. Application of this method in a mouse model of intestinal neoplasia revealed the biogeography of the mouse gut microbiome as function of location in the intestine, frequent strong intermicrobial interactions at short length scales and tumor-associated changes in the architecture of the host–microbiome interface. This method is compatible with widely available commercial platforms for spatial RNA sequencing and can therefore be readily adopted to study the role of short-range, bidirectional host–microbe interactions in microbiome health and disease. Bulk polyadenylation improves the capture of microbial RNA, enabling host–microorganism interactions to be visualized at a high resolution when combined with commercially available spatial transcriptomics platforms.
|
|
Scooped by
mhryu@live.com
Today, 12:23 PM
|
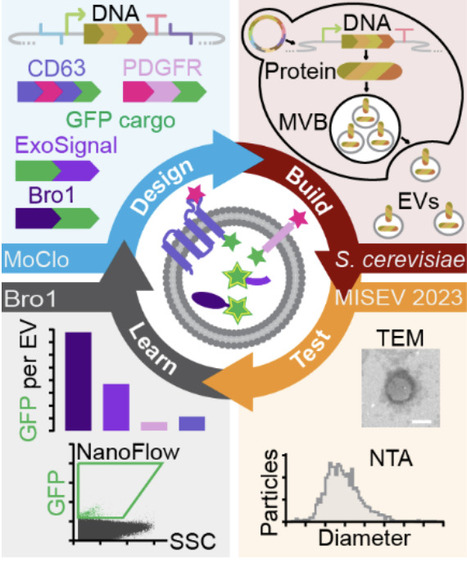
Extracellular vesicles (EVs) hold great promise as therapeutic delivery vehicles, leveraging their natural role as mediators of intercellular communication in all organisms studied. However, many barriers must be overcome to realize their full potential. Saccharomyces cerevisiae is an attractive chassis organism to explore solutions: It is used for drug biomanufacturing, it is amenable to complex genetic engineering, and their EVs can drive responses in human cells. To further develop this prospect, we sought to genetically modify S. cerevisiae EVs by devising a research framework amenable to iterative design, build, test, learn cycles, a core principle of synthetic biology. Using this approach, we focused on identifying new scaffolds, proteins that load cargoes into EVs, from a small pool of candidates. We first optimized a modular cloning strategy, called EVclo, for plasmid and genome-integrated candidate gene expression. Candidate genes were fused to EGFP, and after confirming expression in cells, we showed that scaffold-EFGP proteins colocalized with mRuby2-tagged Nhx1, a biomarker of multivesicular bodies, presumed sites of EV biogenesis. We triggered release of EVs by heat stress, isolated these EVs by ultrafiltration and size exclusion chromatography, and confirmed the presence of exosome-sized EVs in all samples. We find that candidate scaffold proteins did not affect EV size, morphology or titers. Further analysis of these samples indicated that some EGFP-tagged scaffolds are present in EVs: Bro1, a yeast ortholog of ALIX, was most abundant and ExoSignal showed highest enrichment of the human candidates. In all, we conclude that Bro1 is a good scaffold for future engineering strategies, and that human proteins can be sorted into yeast EVs suggesting conservation of the sorting machinery and demonstrating that yeast EVs can be humanized. This synthetic biology-based, proof-of-concept study establishes S. cerevisiae as a platform to engineer and bioproduce designer EVs for many applications.
|
|
Scooped by
mhryu@live.com
Today, 12:16 PM
|
Microbial single-amplified genome (SAG) sequencing technologies have elevated microbial research resolution to the single-cell level. However, neither upstream data processing nor downstream analysis has been fully developed, greatly limiting the research in strain level. Herein, we developed MetaSAG (Multi-level Exploration and Taxonomic Analysis of microbial Single-Amplified Genomes), which enables accurate and rapid taxonomic classification of microbial SAGs. MetaSAG outperforms existing method in species classification certainty, computational efficiency, and sensitivity of low abundance species identification. In addition, MetaSAG enables species-level functional analysis, as well as strain-level evolutionary analysis. With the help of MetaSAG, we discovered the parasitic relationship between phages and bacteria, identifying multiple susceptible bacteria and a broad spectrum of phages. Furthermore, we developed MetaK-Lytic (k-mer-based meta-learning framework to predict phage lytic ability) to achieve accurate prediction of phage lytic activity based on 31-mer short sequences, which is well adapted to the characteristics of incomplete SAG sequences. Overall, we offer a comprehensive integrated tool that can parse microbial SAG data from raw data to the strain level to decipher the functional ecology of microbial dark matter, with broad implications for microbial ecology and phage therapy (https://github.com/liangcheng-hrbmu/MetaSAG).
|
|
Scooped by
mhryu@live.com
Today, 12:00 PM
|
Proteins are built from 20 canonical amino acids. It is interesting to explore whether proteins can be formed from significantly reduced amino acid alphabets. Our bioinformatics survey of UniProt (more than 250 M sequences) revealed that proteins composed of reduced amino acid alphabets (<~10) are extremely rare among existing proteins. Next, we used computational protein design to design proteins composed of all 1,013 possible alphabets of 2-10 early amino acids (Ala, Asp, Glu, Gly, Ile, Leu, Pro, Ser, Thr, and Val). The length of all proteins was 100 amino acid residues. Small amino acid alphabets preferred simple helices or helix bundles. Larger amino acid alphabets allowed for the design of more complex structures. A protein composed of 8 amino acids (Ala, Asp, Gly, Leu, Val, Ser, Thr, and Pro) was successfully experimentally verified. It belongs to fibronectin type III domain beta-sheet-rich architecture. Attempts to experimentally verify designs composed of 6 and 4 amino acids were unsuccessful. We show by a computational experiment without an experimental validation that inverse folding programs, namely ProteinMPNN, can stabilize designed proteins within the same amino acid alphabet. Our results show that globular proteins may have formed early in evolution. Furthermore, we show that it is possible to design proteins with interesting properties for biotechnology and synthetic biology.
|
|
Scooped by
mhryu@live.com
Today, 11:53 AM
|
Drought reshapes plant root microbiota, yet the mechanistic drivers and consequences of this observation remain unclear. We discovered that suppression of host immunity and iron homeostasis is required for Streptomyces enrichment in roots during drought across diverse soils. Genetic and physiological manipulation of these host pathways confirmed their requirement in modulating Streptomyces root enrichment. Drought-induced suppression of iron uptake was conserved across the ~160 My monocot-eudicot divergence. Some Streptomyces strains enhanced plant growth and rescued iron uptake under drought. These benefits were uncoupled from Streptomyces root enrichment. They were instead shaped by intra-Streptomyces antagonism. We propose a two-step model: drought-driven down regulation of host iron and immune pathways enriches Streptomyces, while intra-genus dynamics fine-tune strain-level assembly and functional outcomes. Our data refine the idea that Streptomyces are enriched in roots during drought in response to a plant cry for help and consequently contribute to alleviation of this abiotic stress.
|
|
Scooped by
mhryu@live.com
Today, 11:42 AM
|
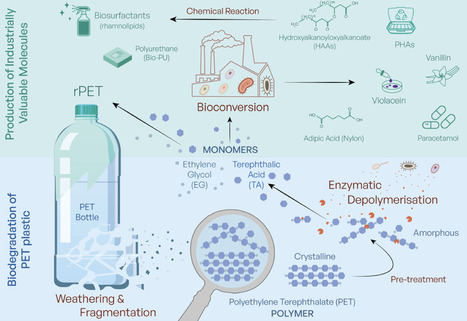
Plastics drive twin crises: persistent pollution and greenhouse gas emissions. Bio-based approaches using enzymes and microorganisms to depolymerise plastics and valorise monomers show promise but raise societal, ethical and regulatory questions central to Responsible Research and Innovation (RRI). In this Perspective, we reflect on RRI implications of bio-based plastic degradation, informed by stakeholder discussions across the plastics value chain and public engagement. We identify broad support alongside concerns about scalability, interaction with existing recycling, governance and containment of genetically modified organisms, management of additives and contaminants, and the roles of regulation and economic incentives in enabling adoption. Bio-based approaches to plastic degradation have been an intense area of research in recent years, and although they show great promise, they also raise societal, ethical and regulatory questions. Here the authors reflect on the Responsible Research and Innovation (RRI) implications of this growing field, sharing insights they have gained through engagement with stakeholders and the broader public.
|
|
Scooped by
mhryu@live.com
March 6, 6:11 PM
|
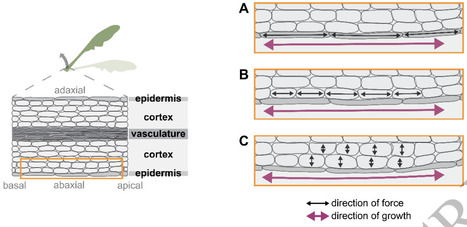
Plants use light both as a resource for photosynthesis and as a signal about their environment. In response to light cues, plants can move their organs via directional growth driven by cell expansion. In dense vegetation where light is available in spatially heterogeneous patterns, plants need to navigate this space to improve the position of their photosynthetic tissues. In canopies blue light irradiance and red to far-red light ratio decrease due to absorption by chloroplasts, and these changes regulate distinct processes within the plant. Changes in light environment are detected by cryptochrome and phytochrome photoreceptors, both regulating Phytochrome Interacting Factors (PIFs) and thereby enhancing elongation in hypocotyls, stems and leaves, and inducing upward leaf movement (hyponasty). An additional class of photoreceptors, phototropins, decode horizontal light gradients to produce directional growth towards the light source (phototropism). Here we review the current state of knowledge on these differential growth responses to light cues, with specific emphasis on the regulatory pathways that translate light signaling in differential cell expansion. Downstream of the photoreceptors, the phytohormone auxin induces cell growth in shoot tissues, but also other phytohormones contribute to balancing light responses. Cell expansion is regulated primarily at the level of cell walls and a comparison of different transcriptome datasets reveals that only a small group of cell wall modifying genes are tightly regulated by shade cues. It remains poorly understood which cell layers are causal to the initiation of cellular expansion. Here we combine insights from different differential growth behaviors in different species and organs to generate different hypotheses for the cellular underpinnings of light-driven leaf movements.
|
|
Scooped by
mhryu@live.com
March 6, 5:04 PM
|
Recent studies offer insights into how the ancient technique works, while newer approaches are bolstering acupuncture with insights from Western medicine.
|
|
Scooped by
mhryu@live.com
March 6, 4:59 PM
|
Spatially regulated membrane constriction is an important milestone in reconstituting minimal cell division. In giant lipid vesicles, bottom-up approaches have reproduced the assembly, mid-cell positioning, and the initial constriction of an FtsZ-based minimal divisome. However, progressive deformation towards giant vesicle scission by near-equatorial Z rings could so far never be observed. One obvious major limitation has been the scale mismatch, as pure reconstituted FtsZ rings typically exhibit bacterial diameters, too small to constrict typical cell-sized vesicles. Therefore, we explore the potential of other key divisome factors to scale up FtsZ-ring functionality in vitro to match the dimensions required for synthetic cell division. We here focus on cytoFtsN, the cytosolic domain of FtsN, and its effect on FtsZ self-organization. Remarkably, a molar excess of cytoFtsN promotes the formation of large, closed equatorial FtsZ rings on giant vesicle membranes, which are able to constrict to almost full closure. By fluorescence imaging and biochemical analysis, we show that cytoFtsN regulates the spatial organization of the FtsZ network primarily by aligning FtsZ filaments while reducing filament depolymerization. Our findings help to define key requirements in a minimal filament-based system for progressive membrane constriction and thus represent a major step forward towards constructing synthetic cells capable of self-division. Spatially regulated membrane constriction is an important milestone in reconstituting minimal cell division. Here the authors engineer a truncated system where the cytosolic domain of FtsN forms large, closed equatorial FtsZ rings on giant vesicle membranes and regulate the FtsZ network formation.
|
|
Scooped by
mhryu@live.com
March 6, 1:30 PM
|
Bio-pesticides constitute a category of natural products and organisms effective against pests. Given their advantages, such as easier clearance and degradation compared to chemical pesticides, along with their lack of detrimental effects on ecosystem health, they represent a promising alternative for sustainable pest management and crop protection. The development of natural biopesticides to substitute synthetic pesticides is therefore of paramount importance for securing food safety and advancing agricultural production. Monoterpenes, primarily present in plant volatile oils, are characterized by a strong aroma and a range of biological activities-including insecticidal, antimicrobial, antiviral, anti-inflammatory, antioxidant, antitumor, and anticancer effects. This profile renders them a promising platform for the research and development of new, more effective pesticides. This article reviews the biosynthetic pathways of monoterpenes and contemporary engineering strategies for their microbial synthesis. Additionally, an evolutionary analysis database for monoterpene synthases (MTPS) has been established. Finally, the biological activities of monoterpenes are summarized and analyzed, with particular emphasis on their potential and advantages as biopesticides, thereby providing direction for the future development of monoterpene-based biopesticides.
|
|
Scooped by
mhryu@live.com
March 6, 1:14 PM
|
The increasing prevalence of antibiotic-resistant bacteria has intensified the demand for novel antimicrobial agents. Antimicrobial peptides (AMP) have emerged as promising alternatives, yet their identification or classification remains challenging due to the lack of multi-perspective information, insufficient feature representation learning, and monocular data modalities. In this paper, we propose a dual diffusion model-based representation learning framework for classifying AMPs, which effectively integrates both peptide sequence and structure information to address existing issues for the task. Specifically, our approach utilizes a multi-view feature construction module, which encodes peptide sequences and structures from distinctive perspectives, deriving initial feature representations with enriched biological semantics. To enhance representation learning, the proposed framework leverages both diffusion models for sequence and structure information respectively to effectively capture complex semantics from dual modalities. In addition, both single-modal and dual-modal contrastive learning are used to further advance the representation learning. Results of comprehensive experiments demonstrate that our model outperforms existing methods for the task of AMPs classification, providing a feasible solution to accelerating the discovery of novel antimicrobial agents.
|

























3st, engineering a POI to control a cell-cycle regulator that is essential for one step of cell proliferation but detrimental to another. if the POI switches between 1-0-1-0-… outputs, driven by repeated cycles of inputs ia-ib-ic-id-…, then cells will proliferate maximally. Any error, either in switching speed or state, e.g., a POI output sequence of 1-1-1-0-…, causes skipped cell cycles, incurring a fitness penalty and giving an advantage to mutants with better fidelity to the desired input-output relationships. we used El222/LIP and transformed yeast with constructs in which LIP controlled the transcription of specific cell-cycle regulators. we deleted cell-cycle control genes in order to make the light-controlled cell-cycle gene essential. to increase the mutation rate, we introduced the pol3-L523D mutation that is reported to increase the global mutation rate by about 100-fold.