Your new post is loading...

|
Scooped by
mhryu@live.com
Today, 1:00 AM
|
The bacterial 16S rRNA gene is widely used to characterize host-associated and environmental microbiomes, most commonly through sequencing short hypervariable regions. Recent improvements in PacBio sequencing chemistry and concatenation approaches can now enable high-throughput, full-length 16S rRNA gene sequencing with high accuracy and depth. However, errors introduced during library preparation remain a major limitation, particularly during PCR amplification of full-length amplicons, where error accumulation may be elevated due to longer sequence lengths. These challenges are amplified when samples vary widely in microbial biomass, making it difficult to select a single optimal number of PCR cycles. Here, we evaluated PCR cycle autonormalization for PacBio Kinnex full-length 16S rRNA gene sequencing across seven agriculturally relevant specimen types. We compared conventional fixed-cycle PCR protocols (20, 24, and 30 cycles) with an autonormalization approach in which individual reactions were terminated during exponential amplification based on real-time fluorescence thresholds. Under the workflow tested here, autonormalized libraries generally retained a high proportion of sequences following denoising and chimera removal, exhibited low residual error rates (<0.005%), and yielded relatively even read distributions across heterogeneous sample inputs. Overamplified reactions (30 cycles) showed elevated residual error rates and greater sequence loss, particularly in samples with higher microbial biodiversity, whereas low-cycle libraries produced more variable read output among specimens. Importantly, the PCR protocol had relatively minor effects on overall community composition compared with specimen type. These results support PCR cycle autonormalization as a useful workflow strategy for heterogeneous full-length 16S library preparation, while also highlighting the importance of library design, pooling strategy, and downstream processing in shaping technical outcomes.
|
|
Scooped by
mhryu@live.com
Today, 12:42 AM
|
RNA polymerase catalyzes transcription, the first step of gene expression. In bacteria, numerous regulatory proteins and signaling molecules fine-tune RNAP activity in a promoter-specific manner. The resultant changes in gene expression allow cells to acclimate to an ever-changing environment. In addition to phenotypic adaptation, increases in cell fitness can also result from changes in the genome. Here, we explore how mutations in RNAP structural genes benefit cells under diverse selection pressures, with a focus on antibiotics. Selection for resistance to rifampicin (RIF), an antibiotic that binds near the catalytic center of RNAP, leads almost exclusively to amino acid substitutions in the large β subunit that modify the RIF binding site. RIFR mutations have pleiotropic effects and can lead to increased or decreased sensitivity to other antibiotics. In addition, mutations in RNAP are linked to resistance to β-lactams, antibiotics that target peptidoglycan synthesis. Mutations in RNAP can act by altering the interaction with key regulators, including the sigma (σ) factors required for promoter recognition, transcription factors, or signaling molecules that bind to RNAP. RNAP mutations also affect catalysis with impacts on promoter recognition and clearance, elongation, and termination. We consider illustrative examples of changes in RNAP that alter the transcriptional landscape to facilitate the emergence of antibiotic tolerance and resistance, both in the laboratory and during the clinical course of treatment in patients.
|
|
Scooped by
mhryu@live.com
June 22, 11:47 PM
|
The twin-arginine translocation (Tat) system is a mechanistically unique protein transport pathway moving folded proteins across membranes. It is found in all domains of life and is essential for bacterial virulence and plant photosynthesis. The membrane proteins, TatA, TatB and TatC form a core complex to which substrate proteins bind, triggering the recruitment of additional TatA protomers to form the transport site. Here we present cryo-electron microscopy structures of the prototypical TatBC complex from E. coli and the atypical complexes from Nitratifactor salsuginis and Myxococcus xanthus in a resting state, alongside TatAC substrate-bound TatBC and TatABC complexes from E. coli in the early stages of transport. These structures demonstrate that substrate proteins associate with the core complex solely through their N-terminal signal peptides. The Tat targeting sequences of the signal peptides make specific contacts with TatC, and the peptide body is clamped by TatB. The core complex contains highly tilted transmembrane helices that drive extreme local membrane thinning. On the basis of our structures and biochemical and functional analyses, we propose a model for the early steps in Tat transport. Cryo-EM structural, biochemical and functional analyses of the bacterial twin-arginine translocation system for protein transport across membranes reveal mechanisms of substrate binding.
|
|
Scooped by
mhryu@live.com
June 22, 11:28 PM
|
Bacterial consortia, with broad metabolism and environmental resilience, show promise for bioaugmentation treatment of plastic wastes. Consortia design principles and plastic degradation enhancements in laboratory and simulated-system studies were reviewed. Practical barriers include narrow polymer scope, long treatment duration, limited scalability, ecological risks, and scarce techno-economic assessments. Bioaugmentation can be realized through the development of bacterial formulations, the integration of pretreatment–bioaugmentation workflows, and the implementation of long-term field trials.
|
|
Scooped by
mhryu@live.com
June 22, 11:23 PM
|
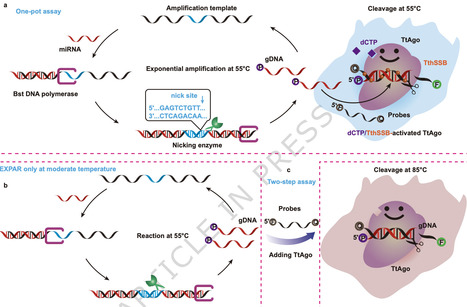
Thermus thermophilus Argonaute (TtAgo) is a DNA-guided endonuclease promising for gene editing and molecular diagnostics. However, its high-temperature dependence (65–85 °C) restricts its utility in moderate-temperature scenarios. Here we show that combining deoxycytidine triphosphate (dCTP) with the single-stranded DNA-binding protein from T. thermophilus (TthSSB) enables robust TtAgo activity within 37–60 °C. Mechanistic studies reveal that dCTP increases TtAgo’s conformational flexibility, while TthSSB promotes substrate recruitment, synergistically driving efficient DNA cleavage. Leveraging this mechanism, we develop ERCB-TtAgo (exponential amplification reaction by dCTP/TthSSB-activated TtAgo), a one-step isothermal diagnostic platform for microRNA detection. It achieves femtomolar sensitivity within 40 min and accurately distinguishes hepatocellular carcinoma patients from controls in a 151-sample clinical cohort, matching RT‑qPCR performance. Our work not only surmounts a key limitation of TtAgo but also offers an approach for modulating nuclease activity, paving the way for intelligent biosensing platforms with broad applicability. Thermus thermophilus Argonaute (TtAgo) requires high temperatures for activity. Here, authors show that dCTP and a single stranded DNA binding protein enable TtAgo to work at moderate temperatures, allowing them to develop an isothermal miRNA detection platform for the diagnosis of hepatocellular carcinoma.
|
|
Scooped by
mhryu@live.com
June 22, 10:37 PM
|
Ammonia-oxidizing archaea (AOA) are widespread and highly abundant in nature. Despite their typical aerobic metabolism, they thrive in ecosystems where oxygen is scarce. Recently, the AOA Nitrosopumilus maritimus SCM1 was shown to produce oxygen and N2O from nitrite upon oxygen depletion, with N2O subsequently converted to dinitrogen (N2) supporting nitric oxide (NO) dismutation as the proposed metabolic pathway. Here, we explore the ability of other ammonia oxidizers, with diverse phylogenetic affiliations and from different environmental settings to produce oxygen via NO-dismutation. We studied three marine AOA, one soil AOA and two soil ammonia-oxidizing bacteria (AOB). Upon oxygen depletion, all strains accumulated oxygen. In incubations of the AOA strains with 15N tracers, two of them, Nitrosopumilus piranensis D3C and Nitrosopumilus adriaticus CCS1, showed transient 46N2O accumulation followed by linear 30N2 production, similar to SCM1, while Nitrosopumilus adriaticus NF5 and Nitrososphaera viennensis EN76 mainly produced 46N2O from nitrite without further N2 accumulation. These findings indicate that oxygen production through NO-dismutation is a common metabolism in cultured representatives of AOA, albeit with physiological variations between different strains such as the reduction of N2O to N2. Comparative genomics did not reveal a distinct putative N₂O reductase in the AOA strains with linear N2 production, suggesting that the observed physiological differences may not directly stem from gene inventory. The finding of oxygen production in several AOA, as well as AOB, suggests that dark oxygen production is a common trait among archaeal and bacterial nitrifiers and adds a potential explanation for their abundance in oxygen-depleted environments.
|
|
Scooped by
mhryu@live.com
June 22, 10:26 PM
|
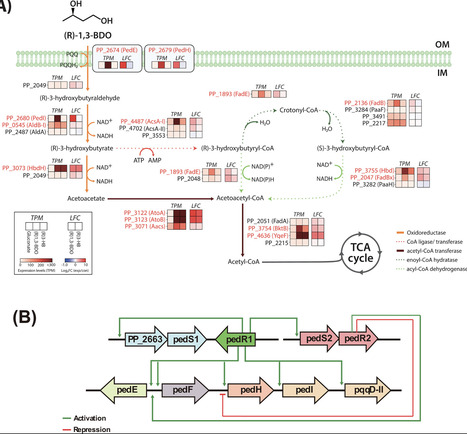
1,3-Butanediol (1,3-BDO) is widely used in consumer and industrial products; however, its microbial degradation remains poorly understood. Here, we dissect the catabolic and regulatory mechanisms of (R)−1,3-BDO utilization in Pseudomonas putida KT2440 and develop (R)−1,3-BDO-responsive transcriptional biosensors. Transcriptomics and qRT-PCR revealed strong induction of the ped gene cluster, which oxidizes (R)−1,3-BDO to (R)−3-hydroxybutyrate [(R)−3-HB], and of the LysR-regulated operon PP_2047–2051, which channels (R)−3-HB toward acetoacetate and acetyl-CoA. Gene deletion and enzyme assays identified pedE and PP_2049 as essential for (R)−1,3-BDO catabolism, with PP_2049 playing a more critical role than the canonical β-hydroxybutyrate dehydrogenase HbdH (PP_3073). Regulatory analysis showed that PedR1 directly activates catabolic genes independently of PedR2—challenging the widely accepted indirect-only model—while PedS1 proved dispensable, implying an alternative sensor kinase. Promoter–GFP fusions demonstrated that full activation of the pedE promoter requires both PedR1 and the PedS2–PedR2 two-component system, whereas the pedS2R2 promoter requires only PedR1 and functions in both native and heterologous hosts, including E coli. These results define the genetic and regulatory architecture of (R)−1,3-BDO degradation in P. putida and establish pedE- and pedS2R2-based promoters as (R)−1,3-BDO-responsive biosensors.
|
|
Scooped by
mhryu@live.com
June 22, 7:29 PM
|
Precise and efficient replacement of large genomic DNA segments without inducing double-strand breaks (DSBs) remains a central challenge in genome engineering. Traditional homologous recombination relies on DSBs and long homologous arms, yet it remains inefficient, while recombinase or integrase systems suffer from residual sequences at integration sites. Prime editing (PE), limited by the processivity of reverse transcriptase, struggles to integrate large fragments (>100 bp). To address this challenge, we introduce Prime Editing–Microhomology-Enabled Replacement (PREMIER), a DSB-free platform by installing single-stranded microhomology arms at donor and genomic junctions via PE. In cell lines, PREMIER achieved a mean efficiency of 63.4% (median 65.2%) in diverse target sites, with peak efficiencies reaching 85.9%, exceeding homology-directed repair by 10–20-fold and reducing off-target integrations by over 100-fold compared to nonhomologous end joining. It bypasses the need for long homology arms, simplifies donor preparation, achieves targeted replacement of sequences up to 10.3 kb. In vivo, PREMIER integrates a 6.2-kb oncogene cassette into the mouse liver. Additionally, PREMIER replaces murine Trp53 with human TP53 CDS, generating functional humanized mice. Altogether, PREMIER provides a precise, high-efficiency, and DSB-free strategy for large-scale genome rewriting, offering a powerful tool for complex modeling and therapeutic genome editing.
|
|
Scooped by
mhryu@live.com
June 22, 7:15 PM
|
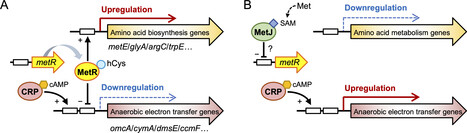
Microorganisms living in nutrient- and energy-limited environments must finely coordinate anabolic and catabolic pathways. However, the molecular mechanisms underlying this balance remain poorly understood. Here, we report a mechanism cross-regulating amino acid biosynthesis and anaerobic respiration in bacterial cells. Using the model bacterium Shewanella oneidensis MR-1, we demonstrated that methionine activates extracellular electron transfer activity at submillimolar concentrations. This regulation is mediated by the transcription factor MetR, a canonical regulator of methionine biosynthesis. Under methionine-limited conditions, MR-1 upregulates methionine biosynthesis genes while repressing genes involved in extracellular electron transfer (metal reduction) and other anaerobic respiratory processes. Conversely, methionine availability relieves this repression, enhancing extracellular electron transfer. Furthermore, the MetR-mediated regulation of extracellular electron transfer was observed in Aeromonas hydrophila. These findings reveal a novel physiological role for MetR, suggesting its involvement in the coordinated energy distribution between anabolic and catabolic pathways.
|
|
Scooped by
mhryu@live.com
June 22, 7:08 PM
|
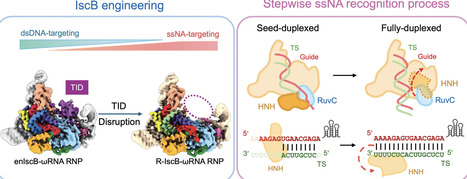
Transposon-encoded IscB was defined as the evolutionary ancestor of CRISPR-Cas9. This compact RNA-guided endonuclease has since been engineered for genome-editing applications. We previously repurposed IscB and related Cas9s as efficient RNA editors by removing their double-stranded DNA (dsDNA) recognition module, the target-adjacent motif (TAM)/protospacer adjacent motif-interacting domain. Here, we report four cryo-electron microscopy structures of IscB, with or without TAM-interaction domain (TID), in complex with single-stranded nucleic acid (ssNA) targets. Structures reveal that, regardless of TID presence, IscB engages ssNA using the same mechanism. IscB initially facilitates formation of a 10-nt seed duplex with ssNA; further base-pairing is blocked by an alternatively positioned HNH nuclease that acts as a roadblock. In this intermediate state, neither HNH nor RuvC is competent for target cleavage. Only upon full duplex formation is the HNH roadblock dislodged by the duplex extension between guide RNA and ssNA. HNH and RuvC nuclease active sites become exposed as the result. A similar set of conformational rearrangements likely governs IscB activity during dsDNA target interrogation. Guided by the structural and mechanistic insights, we introduced mutations to either improve ssNA binding or ease HNH dislodging. Both approaches improved the RNA-targeting efficiency of IscB in vitro and in human cells.
|
|
Scooped by
mhryu@live.com
June 22, 6:43 PM
|
Patients infected with life-threatening multi-drug resistant (MDR) bacteria have been treated with cocktails of bacteriophages. This is a complicated form of personalized medicine as the phages given to a patient have to be selected beforehand on the basis of their lytic capacity of the infecting bacteria. Because bacteria rapidly become resistant, the evolution of resistance to a diverse cocktail of phages is a complicated dynamical process, during which competing bacterial strains replace one another by accumulating several resistance mechanisms, each of which may involve a fitness cost. As a consequence, it is typically not known why a particular phage therapy succeeded or failed, and how one can optimize the composition of the cocktails to maximize the rate of success. To improve upon this, we extend an existing in vivo-calibrated mouse model into a novel mathematical model for the human situation, and include multiple phages infecting multiple bacterial strains, differing in their resistance to each of the phages. We adjust several parameter estimates of the bacterial model to the human situation, and use the model to describe a successful case of phage therapy involving several cocktails, each containing several phages. In the model, treatment success crucially depended on pretreatment resistance levels, and on the diversity and the timing of the cocktails. Once an appropriate cocktail is found, it is less important to further optimize the infection rates of the phages. Resistant bacterial strains expand rapidly when sensitive strains decline, and the higher the infectivity of the phages, the faster resistant strains expand. Because resistance evolves rapidly, it is best to provide a diverse set of phages right from the start of therapy, i.e., to hit hard and early, and create a high genetic barrier to bacterial resistance.
|
|
Scooped by
mhryu@live.com
June 22, 6:31 PM
|
Transcriptional interference is rarely documented in bacteria. In cyanobacteria, phycobilisomes are the major light-harvesting antennae, and their degradation under non-optimal conditions follows a tightly regulated genetic program leading to the production of NblA, a widely conserved proteolytic adapter. NblA production is regulated at both the transcriptional and post-transcriptional levels. Here, we uncover an additional regulatory layer mediated by an antisense RNA conserved in heterocyst-forming cyanobacteria. In Nostoc sp. PCC 7120, transcription of the abundant antisense RNA (as_nblA) limits nblA mRNA accumulation. A strain unable to transcribe as_nblA produces an excess of NblA, ultimately so harmful that suppressor mutations of nblA expression accumulate rapidly, underscoring the essential role of as_nblA. Rifampicin time-series experiments and the inability of as_nblA to regulate nblA expression in trans support transcriptional interference as the primary regulatory mechanism of as_nblA. Mathematical modeling of nblA expression, supported by biological data, shows that as_nblA plays a pivotal role in preventing leaky nblA expression under non-inducing conditions. Our work highlights the importance of antisense RNA-mediated regulation, particularly transcriptional interference, in establishing thresholds that prevent spurious expression of genes encoding critical cellular functions.
|
|
Scooped by
mhryu@live.com
June 22, 5:35 PM
|
Ribosomal proteins contain flexible terminal regions that are averaged out during electron density reconstructions, rendering them absent from experimental models derived by X-ray crystallography or cryogenic electron microscopy. These flexible protein fragments (FPFs) collectively form an invisible coat on the ribosome surface whose presence has been systematically overlooked. Here we analyzed FPFs from 36 ribosomes spanning bacteria, eukaryotes, and mitochondria. We found that mitoribosomes harbor the most numerous and longest FPFs. Structural predictions confirmed that FPFs are predominantly disordered across all ribosome classes. Comparison of FPF amino acid composition against proteome-wide background frequencies revealed strong and domain-specific compositional biases. The balance between arginine and lysine content tracks the cardiolipin content of the membrane each ribosome class contacts. The arginine enrichment in mitoribosomal FPFs may additionally reflect selection arising from the RNA-rich environment of mitochondrial RNA granules, membraneless condensates where mitoribosomes are assembled. FPFs are uniformly depleted in aromatic residues, arguing against protein-driven liquid--liquid phase separation propensity. Our findings suggest that the flexibly tethered coat is a highly functional intrinsic part of all ribosomes.
|
|
|
Scooped by
mhryu@live.com
Today, 12:52 AM
|
The engineered Photorhabdus virulence cassette (PVC) enables precise protein delivery but has not yet achieved RNA packaging and injection delivery. In this study, we achieved intraluminal RNA loading via the U1A RNA-binding domain, anchoring it to the PVC inner tube and establishing DART (PVC Docker-based All-purpose RNA Injection Delivery Tool). This enabled the protective loading of diverse RNAs, including Pepper RNA, guide RNA, siRNA, miRNA, and mRNA. Through the co-delivery of Cas9 in vitro, DART also drove effective knockouts of enhanced green fluorescent protein gene (EGFP), Kirsten rat sarcoma viral oncogene homolog (KRAS), and programmed death-ligand 1 (PD-L1), representing reporter, oncogenic, and immune-related targets for evaluating DART-mediated gene editing. Notably, DART-mediated KRAS knockout produced a significant antitumor effect in a subcutaneous mouse tumor model. Complementary to the external spike-surface fusion strategy of SPEAR (a PVC system termed spike engineering and retargeting), as an intraluminal nanosyringe platform, DART employs internal engineering to expand PVC from protein to RNA delivery.
|
|
Scooped by
mhryu@live.com
Today, 12:38 AM
|
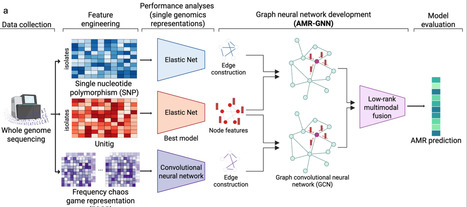
The microbiome actively influences antimicrobial resistance (AMR) dynamics by shaping both ecological and evolutionary processes. However, the extent of its role in resistance emergence, transmission and persistence remains unclear. Traditional AMR research has mainly focused on genetic mechanisms and pathogen-level dynamics. In contrast, the intersection of AMR and the microbiome, including resistance-gene reservoirs, microbial competition and community-mediated selection, remains poorly represented, especially in a modelling context. Here we present a structured framework for incorporating microbiome–AMR interactions into predictive models. We identify key microbiome-mediated processes shaping AMR across different levels of complexity, describe how these can be quantitatively integrated into models, and identify critical data gaps that limit current approaches. By bridging microbiome ecology, AMR biology and mathematical modelling, we set out research priorities and strategies to improve resistance prediction and guide microbiome-targeted interventions. The microbiome plays a significant yet underexplored role in antimicrobial resistance by influencing ecological and evolutionary processes. This Perspective proposes a framework to integrate microbiome–AMR interactions into predictive models while highlighting key mechanisms and data gaps to improve resistance understanding and interventions.
|
|
Scooped by
mhryu@live.com
June 22, 11:37 PM
|
Microbial inoculants are increasingly promoted as sustainable alternatives or complements to conventional agricultural inputs, yet their field performance remains highly variable. This review examines how ecological processes governing root microbiome assembly constrain inoculant establishment, persistence, and function across agricultural systems. We synthesize current evidence on the roles of environmental filtering, host-mediated selection, microbial interactions, and context dependency in shaping inoculant outcomes. We further evaluate the promise and limitations of core-microbiome concepts and synthetic communities as emerging strategies for microbiome-informed inoculant design, emphasizing that their practical translation remains challenged by methodological variability, ecological complexity, formulation constraints, and regulatory barriers. By integrating ecological theory with applied microbiology, this review highlights why many inoculants fail to deliver consistent agronomic benefits and outlines a more potential framework for developing context-aware, field-relevant microbiome-based solutions for sustainable agroecosystem management.
|
|
Scooped by
mhryu@live.com
June 22, 11:25 PM
|
Plastic pollution poses a severe threat to biodiversity and undermines the global ambition of achieving a nature-positive future by 2050. Despite pressing calls for action, the United Nations (UN) treaty on plastic pollution failed to reach consensus at the Intergovernmental Negotiating Committee (INC)-5.1 meeting in Busan (December 2024) and again at INC-5.2 in Geneva (August 2025), where delegates deferred binding commitments. Consequently, biodegradable plastics have gained momentum as a viable alternative, with the global market projected to exceed multi-billion-dollar valuations by 2030, particularly across the Asia-Pacific, Europe, and North America. However, issues of scalability, cost, and mismatches between material performance and waste management infrastructure continue to limit their wider adoption. Although corporate initiatives that prioritize responsible sourcing and waste reduction demonstrate early progress, environmental trade-offs remain. While a global treaty is essential, immediate efforts must address the technical and economic challenges of positioning biodegradable plastics as effective and practical transitional tools. Strategic collaboration between policymakers and industry could accelerate progress toward the UN Sustainable Development Goals, aligning with the 2025 World Environment Day theme of ending plastic pollution. Bridging these gaps offers a promising pathway for mitigating ecological damage as global governance frameworks continue to evolve. Plastic pollution and the increasing economic burden of plastic waste management highlight the urgent need for circular and sustainable solutions. Although bioplastics currently represent less than 1% of global plastic production, their production is rapidly increasing, positioning them as a promising alternative to conventional plastics. Since many bioplastics can be produced from renewable resources, including agricultural residues and other bio-waste, they also offer an effective pathway for waste valorization and sustainable resource management. To further advance the bioplastics industry, greater public awareness, continued research and development, supportive policies, and strong collaboration among academia, industry, government, and other stakeholders are essential.
|
|
Scooped by
mhryu@live.com
June 22, 11:12 PM
|
Xylanases are central to lignocellulosic biomass degradation, yet current methods lack the specificity to resolve how enzymes distinguish complex xylan structures decorated with arabinofuranose (Araf) and 4-O-methyl-glucuronic acid (MeGlcA). Here, we report a suite of chemically-defined activity-based probes (ABPs) that enable the selective detection of arabinoxylan- and glucuronoxylan-specific xylanases (AXXs and GXXs). These cyclophellitol-derived ABPs covalently label retaining xylanases at their active sites, allowing precise mapping of substrate specificity across diverse glycoside hydrolase families. Crystallographic and mass spectrometric analyses reveal the molecular basis of probe selectivity, while in-gel and pull-down assays demonstrate their effectiveness in profiling xylanase activities in complex bacterial and fungal proteomes, including cellulosomes. By integrating activity-based protein profiling (ABPP) with sequence similarity networks (SSNs), we further show that xylanase specificity can be predicted from sequence alone, enabling rapid functional annotation of uncharacterized xylanases. This chemoproteomic strategy provides a powerful platform for discovering and engineering substrate-specific enzymes for biomass valorisation, microbial ecology, and biotechnological applications. Xylan is a complex plant polysaccharide with chemical decorations that shape its degradation. Here, the authors develop selective probes for xylanases targeting these structures in biological samples, offering a powerful approach for enzyme discovery and optimization for biotechnology.
|
|
Scooped by
mhryu@live.com
June 22, 10:29 PM
|
Nicotinamide adenine dinucleotide (NAD+) is one of the most important metabolic coenzymes that not only drives redox reactions and energy production but also acts as a critical substrate for several enzymes involved in immune signalling, DNA repair and epigenetic regulation. Viral infections are known as potent modulators of NAD+ metabolism, with pathogens such as SARS-CoV-2, influenza A virus, Zika virus, herpes simplex virus and human immunodeficiency virus altering NAD+ biosynthesis and consumption to benefit their persistence and replication. In this review, we summarize the current understanding of NAD+ metabolism and its regulatory enzymes: sirtuins, poly (ADP-ribose) polymerases and CD38/CD157. We then discuss the interplay between NAD+ homeostasis and virus infection. Understanding how diverse viruses manipulate NAD+ metabolism could lead to broad-spectrum antiviral strategies grounded in metabolic resilience.
|
|
Scooped by
mhryu@live.com
June 22, 10:22 PM
|
In plant synthetic biology, precise control of gene expression requires constitutive promoters with quantitatively defined transcriptional strengths. Here, we identified endogenous constitutive promoter candidates from Nicotiana benthamiana. The transcriptional activities of the selected promoters were measured using GFP-based and dual-luciferase reporter assays in plants and benchmarked against the CaMV 35S promoter, revealing substantial variation in promoter strengths. Promoter activities were further evaluated in heterologous species, including Brassica rapa, Capsicum annuum, and Zea mays, using protoplast-based dual-luciferase assays. Cross-species analyses showed that relative promoter ranking was partially retained among dicot species, whereas transcriptional activity varied substantially depending on species and was generally reduced in maize. Together, this study defines a set of endogenous constitutive promoters across a range of transcriptional strengths, providing quantitative guidance for selecting endogenous alternatives to the CaMV 35S promoter in plant biotechnology and synthetic biology applications.
|
|
Scooped by
mhryu@live.com
June 22, 7:20 PM
|
Accurate RNA splicing is essential for gene expression and protein function, yet the mechanisms governing splice site recognition remain incompletely understood. Aberrant splicing caused by mutations can lead to severe diseases, including cancer and genetic disorders, underscoring the need for accurate computational tools to predict splice sites and detect disruptions. Existing methods have made significant advances in splice site prediction but are often limited in handling long-range dependencies due to high computational costs, a factor critical to splicing regulation. Moreover, many models lack interpretability, hindering efforts to elucidate the underlying biological mechanisms. Here, we present SpliceSelectNet (SSNet), a hierarchical Transformer-based deep learning model that predicts splice sites from DNA sequences spanning up to 100 kb. By integrating local and global attention mechanisms, SSNet efficiently captures both proximal and distal regulatory signals while maintaining single-nucleotide resolution. Across multiple benchmark datasets, SSNet achieves state-of-the-art performance in splice site prediction and aberrant splicing detection. Systematic in silico mutagenesis demonstrates that attention scores reflect functional sequence importance, supporting their biological relevance. Long-range sequence perturbation experiments further show that SSNet captures distal regulatory effects beyond conventional receptive fields. Together, these results establish SSNet as a biologically interpretable framework for modeling long-range splicing regulation from genomic sequence.
|
|
Scooped by
mhryu@live.com
June 22, 7:12 PM
|
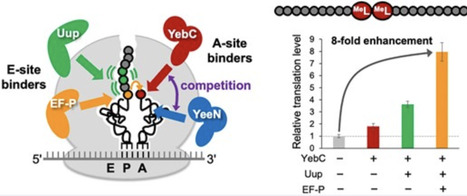
Development of genetic code reprogramming methodologies has enabled the ribosomal incorporation of diverse backbone-altering nonproteinogenic amino acids (BAAs)—including d-α-amino acids, β-amino acids, and N-methyl-α-amino acids—into nascent peptides. However, their incorporation is far less efficient than that of canonical l-α-amino acids. We previously demonstrated that the ribosomal E-site-binding translation factors, EF-P and ABC-F proteins, can enhance BAA incorporation. These findings motivated us to investigate additional ribosome-binding factors that might further facilitate this process. Here, we focused on two putative A-site-binding factors, YebC and YeeN, to activate A-site BAA-tRNA and facilitate efficient incorporation. When testing 11 BAAs for consecutive incorporation, YebC and YeeN yielded average enhancements of 2.1- and 2.7-fold, respectively, with an increase of up to 6.6-fold in specific cases. Furthermore, combining YebC with EF-P and Uup (a representative ABC-F protein) produced an 8.0-fold increase in the incorporation of two consecutive N-methyl-l-leucine residues, demonstrating a clear synergistic benefit. We also found that YebC and YeeN promote ribosomal synthesis of drug-like macrocyclic peptides enriched with BAAs, such as d-serine, 1-aminocyclobutane-1-carboxylic acid, N-methyl-l-alanine, and β3-homomethionine. These results pave the way for the ribosomal synthesis of diverse macrocyclic peptide libraries and their application in mRNA display-based screening for novel bioactive compounds.
|
|
Scooped by
mhryu@live.com
June 22, 6:46 PM
|
How the twin-arginine translocase (Tat) system transports fully folded substrate proteins across cellular membranes without disrupting membrane integrity has been a fundamental question in cell biology for decades. The Tat system, found in prokaryotes and plant organelles, recognizes a cargo signal peptide via a conserved twin-arginine motif. The multi-subunit Tat complex facilitates the proton-motive-force-dependent translocation process, yet its overall architecture has remained unknown. Here, we present the cryo-electron microscopy (cryo-EM) structure of the E. coli trimeric TatB₃C₃ complex with bound substrate SufI, assembled in vivo. The complex adopts an unusual, wide-open, bowl-shaped architecture with a polar inner cavity. Unexpectedly, the cargo is engaged in a dual-contact mode: while the signal peptide binds inside one TatBC unit, the folded domain docks tightly onto an adjacent unit, possibly performing a proofreading function. This structure provides a mechanistic framework for substrate engagement and suggests the direct involvement of the entire Tat complex in substrate translocation.
|
|
Scooped by
mhryu@live.com
June 22, 6:35 PM
|
In this paper we present CoDaLoMic, an R package for analyzing longitudinal and compositional microbiome datasets. The CoDaLoMic package implements three models specifically designed for the analysis of microbiome data that are both compositional and longitudinal. Unlike many existing methods that focus solely on pairwise interactions, CoDaLoMic also captures interactions among groups of bacteria, providing a more robust methodological framework for studying microbial relationships at the community level. In addition, the package facilitates the analysis of microbiome variability in relation to host health status and allows for the identification of groups of taxa that exhibit similar temporal dynamics. Working with time series data makes it possible to understand not only the current state of a microbial community but also its dynamics over time, which is essential for identifying patterns of ecological succession, detecting events of dysbiosis or recovery, and inferring potential causal relationships between taxa. On the other hand, focusing on interactions among groups of bacteria, rather than analyzing only pairwise relationships, enables a more integrated and functionally meaningful view of the microbiome. Many key ecological functions are the result of the collective behavior of functionally related groups of taxa. Two datasets have been considered in CoDaLoMic, one real and one simulated. The real dataset contains the information of the genera present in the microbiome of the Blatella germanica cockroach at 105 time points. The simulated dataset is defined taking Lotka-Volterra structure into account. CoDaLoMic is available at CRAN.
|
|
Scooped by
mhryu@live.com
June 22, 5:44 PM
|
Biofilms are a form of microbial growth consisting of cells, often attached to a surface, embedded in a structured 3D extracellular matrix that confers important emergent properties such as increased resistance to physical removal and antimicrobials. Despite the importance of biofilms to a variety of systems and despite increasing attention from both the public and private sectors, high-throughput approaches to study them are scarce, limiting investigations of complex mechanisms critical for the structure and function of biofilms, such as interactions in multispecies communities. We thus developed a novel workflow to grow and analyze bacterial cells adhered to plastic beads encapsulated within highly parallel nanoliter-scale water-in-oil microfluidic droplets. We term this pipeline for bead-in-droplet biofilm cultivation and characterization BiDBiC. To benchmark BiDBiC, we utilized a well-characterized biofilm former, Stenotrophomonas maltophilia, as well as a poorly studied drinking water biofilm isolate, Sphingopyxis sp. OPL5. Each bacterium exhibited strong adherent growth when co-encapsulated with polystyrene beads in droplets. Furthermore, we retrieved beads from the droplets and removed planktonic cells, enabling focused analysis of adhered cells. From bead-associated biomass, we extracted DNA and RNA for molecular analysis and recovered viable cells for subculturing. We conclude with a discussion of further development of the platform as well as suggestions for microbial biofilm systems that may benefit from ultra-high-throughput droplet-enabled cultivation and analysis.
|