 Your new post is loading...

|
Scooped by
mhryu@live.com
Today, 1:33 PM
|
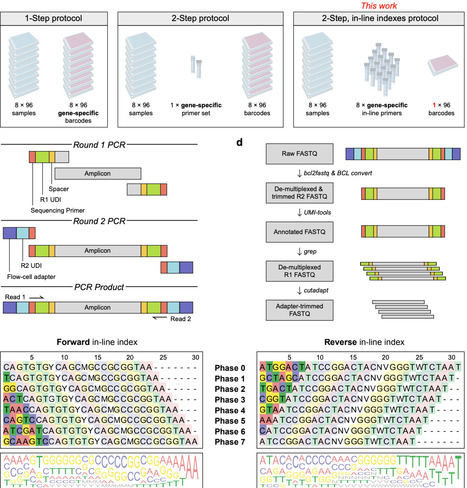
Targeted amplicon sequencing is widely used to profile genetic variation in defined genomic regions. In microbial ecology, for example, amplicon sequencing of the 16S and 18S ribosomal RNA genes has been transformative for characterizing microbial communities. However, on high-capacity sequencing platforms with patterned flow cells, throughput is constrained by the requirement for unique dual indexes (UDIs), which increases primer costs and limits the number of samples that can be pooled per sequencing run. Here, we introduce CUPID-seq (Combinatorial, Unique, Phased, In-line Dual-indexed sequencing), a highly multiplexed amplicon-sequencing strategy that increases scalability through combinatorial indexing across two rounds of PCR. CUPID-seq introduces phased, in-line UDIs during Round 1 gene-specific amplification, enabling multiple samples to share the same Illumina UDI during Round 2 PCR while remaining uniquely identifiable. This design reduces upfront costs by up to 85% and reduces library preparation time and reagent use by up to 40%. We develop and validate CUPID-seq primers targeting the 16S V4 region and provide a computational workflow for demultiplexing in-line indexes. Although optimized here for 16S-based profiling, CUPID-seq can be readily adapted to other user-defined amplicons. By reducing cost and increasing multiplexing capacity, CUPID-seq enables users to leverage high-throughput sequencing platforms more effectively across diverse biological contexts.
|
|
Scooped by
mhryu@live.com
Today, 1:14 AM
|
Consortia of archaea and partner bacteria couple the anaerobic oxidation of alkanes to sulfate reduction. While catabolic pathways in anaerobic alkane-oxidizing archaea (ANKA) are increasingly understood, their anabolic capacities remain poorly characterized. Here, we examined nine enrichment cultures dominated by ANKA and their partner bacteria for small-molecular compounds using solvent extraction and gas chromatographic analysis of derivatized extracts. All hydrocarbon-degrading cultures contained substantial amounts of disaccharides in their metabolite pools. Cold-adapted methane-oxidizing cultures dominated by ANME-2c and Seep-SRB2 contained up to 1.5 mg of trehalose per mg soluble protein. Trehalose was also abundant in ethane-oxidizing cultures of Candidatus Ethanoperedens and its distinct partner SRBs, accounting for up to 75 % of the extracted metabolites. In contrast, thermophilic ANKA cultures dominated by ANME-1 or Ca. Syntropharchaeum and Ca. Desulfofervidus contained an abundant as-yet-unidentified glucose-containing disaccharide. Metagenomic analysis revealed widespread trehalose metabolism genes among partner Desulfobacterota and in ANME-2c and Ca. Ethanoperedens, but a lower potential in ANME-1 and Syntropharchaeum, consistent with metabolite profiles. If exogenous trehalose was added to the Ethane50 culture, we observed rapid metabolization by heterotrophic microorganisms, but poor assimilation by the Ca. Ethanoperedens/ Ca. Desulfofervidus core community, indicating that ANKA/SRB consortia do not consume externally supplied trehalose. Instead, Ca. Ethanoperedens/ Ca. Desulfofervidus, as well as other ANKA/SRB consortia, may use the disaccharides as energy-storage molecules, osmolytes, or components of the extracellular matrix. Notably, the disaccharides produced by the consortia also sustain ancillary heterotrophs, thereby linking alkane oxidation to broader sedimentary carbon cycling.
|
|
Scooped by
mhryu@live.com
Today, 12:50 AM
|
Surface display of proteins on bacteria bears several advantages for binding studies, library screening or enzyme inhibitor testing. Here, we present a set of plasmids for autodisplay-based surface expression of recombinant proteins, named Autodisplay-ToolBox (ATB). In this set, crucial parts as required for autodisplay, including promotor, SP, linker and β-barrel can be combined in varying permutations to find the best combination for a certain protein. The plasmids can be applied individually or in library approaches, enabling optimization of surface display in a single step. By such a library approach, the activity of surface-displayed β-glucosidase (β-Gluc) is increased by a factor of 4.9, the activity of a laccase (CotA) by a factor of 4.7 and the binding capacity of the surface-displayed nucleotide binding domain of human HCN2 channel (HCN2-CNBD) by a factor of 10.3. It is shown that the ATB can be used in different strains of E. coli as well as in Pseudomonas putida. The aim is to provide a selection of plasmids, accessible by Addgene, that every scientist can use for their own protein, either individually based on personal preferences, structural features, etc., or as a library, as shown here for three different examples. A toolbox of 81 plasmids, accessible by Addgene, facilitates tailor-made autodisplay of proteins in bacteria, e.g. E. coli or P. putida. Plasmids can be applied individually or as a library for optimal promotor, SP, linker, and β-barrel combination.
|
|
Scooped by
mhryu@live.com
Today, 12:32 AM
|
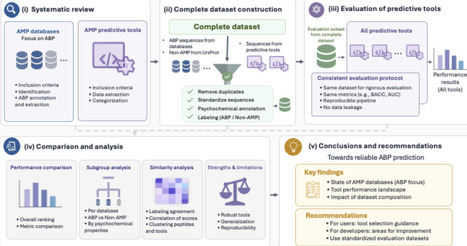
Antimicrobial resistance (AMR) has a profound impact on animal and human health and is associated with substantial morbidity, mortality and public health costs. There is a clear need to develop novel, effective antibiotic agents, which can overcome the current AMR crisis. Antimicrobial peptides (AMPs) may offer such a solution and have attracted growing attention for their potential to combat AMR. In parallel, the growing availability of peptide sequences in public databases has stimulated the development of numerous machine learning and deep learning tools to predict antimicrobial activity computationally. However, it remains unclear how reliably these tools can be compared, as existing studies often rely on heterogeneous datasets and inconsistent evaluation protocols that may lead to data leakage and inflated performance estimates. This raises a central question: what evaluation criteria and benchmark resources are needed to enable fair, reproducible, and biologically meaningful assessment of AMP prediction tools? We address this question by focusing specifically on antibacterial peptides (ABPs). We first provide an overview of AMP databases relevant to antibacterial activity and compare their content, redundancy, and experimental metadata. We then critically assess existing computational tools for ABP prediction, highlighting key limitations related to dataset construction, affinity to certain sequences, data leakage, and inconsistent performance reporting. Based on these limitations, we propose a reference evaluation framework designed to improve comparability, reproducibility, and practical utility in ABP prediction. Finally, we provide targeted recommendations for AMP databases and future tool development to support more robust progress in the computational discovery of ABPs.
|
|
Scooped by
mhryu@live.com
Today, 12:20 AM
|
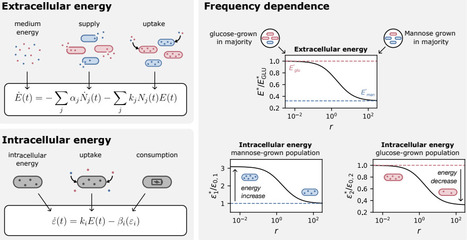
Bacterial communities often spend long periods under starvation, where survival depends not only on their intrinsic ability to withstand stress but also on nutrients released by dying neighbors. This creates a distinct form of competition: cells compete for recycled necromass, and the outcome should depend on physiological traits that determine nutrient uptake and maintenance demand. Here, we develop a quantitative framework for this competition using E. coli populations whose starvation physiology is tuned by prior growth history. Fast-grown populations have higher maintenance demands and die slightly faster in monoculture, whereas slow-grown populations are better adapted to starvation. In co-culture, these physiological differences are strongly amplified in a frequency-dependent manner: less-adapted populations die several-fold faster than in monoculture, whereas well-adapted populations can reduce their death rate below that of stationary-phase adapted monocultures. We explain these dynamics with a shared-energy-pool model in which death releases recyclable nutrients, surviving cells consume them for maintenance, and intracellular energy sets death rate. Using independently measured parameters, the model makes parameter-free predictions for competitive survival. The predicted instantaneous death rates collapse onto a universal function of the population ratio over four orders of magnitude. Our results establish necromass recycling as a quantitative basis for bacterial competition during starvation and lay the foundation for modeling communities during starvation.
|
|
Scooped by
mhryu@live.com
Today, 12:01 AM
|
Coexisting strains of the same species within metagenomic data pose a substantial challenge to inferring transmission of pathogenic and commensal microbes. Here we present TRAnsmission Clustering of Strains (TRACS), a highly accurate algorithm for estimating genetic distances between strains at the level of individual single nucleotide polymorphisms, which is robust to intra-species diversity within the host. Analysis of faecal microbiota transplantation datasets and extensive simulations demonstrates that TRACS outperforms existing methods. We use TRACS to infer transmission networks in patients colonized with multiple strains, including severe acute respiratory syndrome coronavirus 2 amplicon sequencing data, deep population sequencing data of Streptococcus pneumoniae and single-cell genome sequencing data from patients infected with Plasmodium falciparum. Applying TRACS to gut metagenomic samples from a mother–infant cohort revealed species-specific transmission rates and identified increased the persistence of Bifidobacterium breve in infants, a finding previously missed owing to the presence of multiple strains. Our study shows that TRACS can be used across microbial kingdoms to uncover strain dynamics. The TRACS algorithm tracks bacteria, viruses and parasites in metagenomic or population sequencing data, detecting microbial transmission even when multiple strains coexist within a host.
|
|
Scooped by
mhryu@live.com
May 21, 11:20 PM
|
G protein-coupled receptors (GPCRs) play key roles in physiology and are central targets for drug discovery and development, but the design of protein agonists and antagonists has been challenging as GPCRs are integral membrane proteins and conformationally dynamic. Here we describe computational de novo design methods and a high-throughput “receptor diversion” microscopy-based screen for generating GPCR binding miniproteins with high affinity, potency and selectivity. We design miniprotein agonists that activate receptors involved in itch and pain, as well as antagonists that inhibit receptors implicated in cancer, metabolic disorders such as diabetes and obesity, and migraine. Cryo-electron microscopy (cryo-EM) structures of five receptor-bound designs are close to the computational design models. A designed chemokine receptor antagonist mobilizes hematopoietic stem and progenitor cells in vivo at a level comparable to a clinically used drug, with fewer adverse effects.
|
|
Scooped by
mhryu@live.com
May 21, 6:08 PM
|
Antimicrobial resistance remains a significant global threat to human health, but microorganisms have long been a crucial source of novel antibiotics. The widely distributed gram-positive bacterium Bacillus subtilis produces an abundance of secondary metabolites, and their antibacterial activities could have significant applications in food, agriculture, and aquaculture areas. These secondary metabolites exert antibacterial effects through mechanisms such as microbial cell membrane structure disruption, cell wall synthesis interference, and cellular metabolic activity inhibition. In contrast to microorganisms such as Streptomyces, B. subtilis forms characteristic biofilms and exhibits quorum sensing, which play important roles in the production of secondary metabolites and their antimicrobial effects. However, limited attention has been focused on the unique molecular mechanisms associated with biofilms and quorum sensing. In this review, we first summarize the typical secondary metabolites produced by B. subtilis. We then mainly focus on the molecular mechanisms associated with the regulation of biofilms and quorum sensing by antimicrobial secondary metabolites, and the effects of biofilms and quorum sensing on the biosynthesis of antimicrobial secondary metabolites. The applications of antimicrobial secondary metabolites in the fields of food, agriculture, and fisheries, based on the regulation of biofilm and quorum sensing, are also summarized. Finally, we highlight the need for further research into the regulatory networks related to biofilms, quorum sensing, and metabolites to facilitate a deeper understanding of the antimicrobial properties of B. subtilis, which may provide theoretical support for the development of novel antimicrobial food technologies.
|
|
Scooped by
mhryu@live.com
May 21, 5:57 PM
|
Orfamide A, a lipopeptide produced by Pseudomonas protegens Pf-5, is a key determinant of its biocontrol properties. In this study, we investigated the regulatory interactions among the GacS/A two-component system, small RNAs (sRNAs), repressor proteins, and two LuxR-type transcription factors in orfamide A biosynthesis. We found that GacS/A indirectly regulates orfamide A production by enhancing transcription of three sRNAs (RsmX, RsmY, and RsmZ). RsmY and RsmZ synergistically relieve repression by RsmA and RsmE, while RsmX plays a lesser role, likely counteracting only one repressor. LuxR-type transcription factors, LuxR1 and LuxR2, which positively regulate orfamide A synthesis, are directly repressed by RsmA and RsmE via binding to their 5′ untranslated regions, linking them to the Gac–Rsm signaling cascade. We further demonstrated that LuxR2 activates luxR1 expression, which in turn facilitates orfamide A production by binding to the promoter of the orfamide A biosynthetic gene cluster. Importantly, we showed that this entire regulatory cascade operates in the rhizosphere and directly influence biocontrol efficacy. These findings provide a comprehensive understanding of the Gac–Rsm–LuxR pathway in orfamide A biosynthesis and offer valuable insights for the development of biocontrol agents based on Pseudomonas strains.
|
|
Scooped by
mhryu@live.com
May 21, 5:48 PM
|
The rise in antimicrobial resistance underscores the need for innovative strategies to combat gastrointestinal infections. Probiotics such as E. coli Nissle 1917 (EcN) offer promising options, but the molecular mechanisms underlying their protective effects remain unclear. We introduce a G66R point mutation in FimH, creating a high-binding EcN variant that more effectively prevents Salmonella Typhimurium attachment and induces a distinct host transcriptional profile, shifting toward adaptive rather than innate inflammatory signaling. In vivo, EcNG66R pretreatment significantly reduced intestinal colonization, fecal shedding, and systemic spread, and prevented splenic enlargement compared with EcNWT. Protection was associated with a marked expansion of CD4+ and CD8+ T cells, essential for clearing intracellular pathogens. EcNG66R further enhanced “readiness” in the spleen under non-infected conditions, without adverse effects on host physiology. EcNG66R thus functions as a dual-action probiotic—improving competitive exclusion while priming cytotoxic T-cell-mediated protection—and provides a promising platform for developing next-generation microbe-based therapies.
|
|
Scooped by
mhryu@live.com
May 21, 5:30 PM
|
The twin-arginine translocation (Tat) system is the only general pathway for the transport of folded proteins across energized biological membranes. It is found in the bacterial or archaeal cytoplasmic membrane, the plant thylakoid membrane or the inner membrane of plant mitochondria. The biological importance of this translocation system can be exemplified by the fact that all bacterial or plant photosynthesis and photosynthetic oxygen evolution on earth requires this system. Despite many biochemical and biophysical studies, the Tat mechanism has been puzzling since the system was discovered in the 1990ies. Important characteristics of the Tat system could not be explained, and also recent high resolution structures of the Tat system’s core with bound substrate has not led to a general transport mechanism yet. In this integrative review, we attempted to answer the key open questions relevant to the Tat mechanism and thereby developed an in its molecular detail new comprehensive explanation of how folded proteins are translocated across membranes by the Tat system.
|
|
Scooped by
mhryu@live.com
May 21, 5:23 PM
|
Virus-like particles (VLPs) are widely used as noninfectious platforms for vaccines, drug delivery, and synthetic biology. Here we report a bottom-up approach to generate liposomal VLPs via post-insertion of protein–lipid conjugates into preformed liposomes. Proteins were covalently coupled to a benzylguanine (BG) lipid via the bioorthogonal and irreversible SNAP-tag/BG reaction, and the resulting protein–lipid conjugate enabled detergent-free insertion into liposomal lipid bilayers. Using a SNAP-tagged GFP conjugate, we first confirmed membrane association by liposome cosedimentation and detected increased GFP fluorescence on individual liposomes by flow cytometry. We next applied the method to the SARS-CoV-2 Spike ectodomain. A Spike-SNAP recombinant protein was expressed in Expi293F cells, and its conjugation capability with BG lipid was verified by a binding inhibition assay using a fluorescent BG substrate. Incubation of virus-sized liposomes with Spike–lipid conjugates resulted in lipid-anchor-dependent recovery of Spike in the liposome fraction and increased Spike-associated fluorescence by flow cytometry without detectable changes in particle size. Negative-stain transmission electron microscopy showed a virus-like texture on Spike-modified liposomes compared with unmodified liposomes. In an ACE2 plate-binding assay, Spike-modified liposomes showed higher binding ability than unmodified liposomes, and the binding was reduced by Congo Red, a reported inhibitor of the Spike–ACE2 interaction, and a neutralizing anti-Spike antibody. By decoupling the design of the conjugate from that of the liposome, this post-insertion approach can facilitate rapid and systematic optimization of VLPs for mechanistic investigations and application-oriented studies.
|
|
Scooped by
mhryu@live.com
May 21, 4:43 PM
|
Oncolytic bacteria have emerged as a promising platform for targeted cancer therapy owing to their intrinsic ability to preferentially colonize tumor tissues, induce direct tumor cell killing, and remodel the tumor microenvironment to activate antitumor immunity. However, native bacteria alone rarely meet the requirements of precision oncology, particularly in terms of spatial specificity, temporal control, and safety. Recent advances in synthetic biology have enabled the construction of stimulus-responsive gene circuits that confer programmable control over therapeutic gene expression in tumor-colonizing bacteria by coupling defined exogenous triggers or endogenous tumor-associated cues to tightly regulated genetic programs. These engineered systems support the tumor-specific delivery of diverse therapeutic payloads, including cytotoxic agents, cytokines, immunomodulatory ligands, prodrug-converting enzymes, metabolic modulators, and nucleic acid-based therapeutics, while minimizing off-target activity. This review thus summarizes recent developments in stimulus-responsive oncolytic bacteria, highlights key design principles and performance trade-offs, and discusses emerging strategies to advance bacteria as programmable living therapeutics for cancer treatment.
|
|
|
Scooped by
mhryu@live.com
Today, 1:20 AM
|
Microbes are increasingly used as living therapeutics, yet their uncontrolled dissemination in the body has remained a clinical roadblock. Physical containment remains largely unattainable owing to eventual bacteria escape. In this work, we present an implantable material that encapsulates and confines bacteria, wherein synthetically engineered microbes produce therapeutic payloads from within. We developed a hydrogel scaffold with dual mechanical features: high stiffness to regulate bacterial proliferation and high toughness to resist material fracture under physiological stress. This design achieved complete bacterial containment for 6 months and withstood multiple forms of mechanical loading that otherwise caused catastrophic material failure. By genetically engineering embedded bacteria, we endowed the material with environmental sensing and on-demand therapeutic release capabilities and demonstrated autonomous treatment in a murine prosthetic joint infection model.
|
|
Scooped by
mhryu@live.com
Today, 1:09 AM
|
Genomes constantly face threats from transposable elements (TEs) and other genomic parasites. While the silencing of existing TE has been well studied, little is known about how cells recognize new invading TEs that they have not previously encountered. Here we explore this question by inserting foreign sequences into S. pombe. Our data revealed that the newly invading TE tj1 is recognized and targeted for silencing by RNAi and heterochromatin. The efficiency of recognition, as well as the degree and stability of silencing, depends on the copy number and insertion location of the TE. We demonstrated that RNA, rather than DNA, is sensed, and that the efficiency of TE recognition correlates with levels of RNA antisense to the TE, generated from upstream transcripts. We also show that various genes of non-transposable nature can initiate silencing. Our data show that silencing may not require recognition of specific elements in the transposon by the host defense systems, and suggest that disruption of host transcription patterns triggers recognition of TE. Genomes must defend against transposable elements, but how cells detect newly invading elements remains poorly understood. Here, the authors show that a newly invading transposon in S. pombe is recognized and silenced via RNAi and heterochromatin through RNA-based sensing mechanism.
|
|
Scooped by
mhryu@live.com
Today, 12:45 AM
|
For microbial functional lipids, industrialization is constrained not by titer alone, but by whether production remains stable during propagation and is scalable in bioreactor operation. Strain stability, dedicated bioreactor design, and fermentation scale-up parameter optimization should be treated as core design criteria from the outset.
|
|
Scooped by
mhryu@live.com
Today, 12:29 AM
|
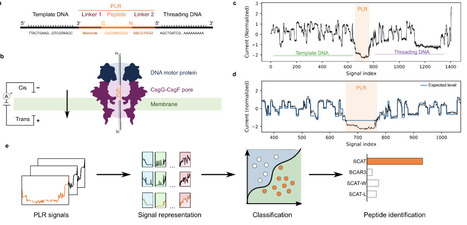
Direct single-molecule peptide analysis could in principle enable rapid and sensitive identification of pathogen-derived or disease-associated biomarkers without reliance on mass spectrometry. However, existing nanopore peptide sensing methods are typically constrained by limited throughput and lack of accessibility beyond specialized setups. Here, we present an integrated experimental-computational framework for DNA-linked peptide translocation on a commercially available, high-throughput nanopore sequencing platform, the MinION. Synthetic peptides were covalently bound to oligonucleotides at both termini. The resulting peptide-DNA constructs were then translocated through the CsgG-CsgF pores using a DNA motor protein. Current traces were segmented using the known DNA sequences to extract peptide-associated signal regions. From these segments, we extracted signal features and trained feature-based and deep-learning classifiers to distinguish peptides, balancing interpretability and classification performance. We establish a framework for peptide identification using standard nanopore sequencing hardware. Across a diverse panel of synthetic peptides, our approach resolves single-amino-acid substitutions, maintains performance across independent sequencing runs, and correctly identifies peptides in blind mixtures. Interpretable model analyses connect classifier decisions and common errors to specific signal motifs. By combining commercially available instrumentation with a reproducible experimental and computational workflow, this framework lowers the barrier to nanopore-based proteomics and enables broader adoption across laboratories. It provides a foundation for future developments in amino acid modification detection and sequence analysis.
|
|
Scooped by
mhryu@live.com
Today, 12:09 AM
|
Generative DNA models are typically next-token completers: they extend a sequence but offer no native interface for telling the model what to make. PlasmidLM is a promptable DNA language model for plasmids. A designer supplies a human-readable component specification, for example a high-copy E.~coli vector with kanamycin resistance and an EGFP reporter, and the model generates the corresponding multi-kilobase construct in a single autoregressive pass. Prompts are unordered sets of named-part tokens at the granularity of biological shorthand, not learned latent codes or rigid grammars. We evaluate outputs along two axes: a sequence is viable if structurally plausible as a plasmid, and faithful if its detected components match the prompt. Their conjunction is the useful-plasmid rate, the primary metric we report. On a held-out 1,000-prompt benchmark, the post-trained model achieves a useful-plasmid rate of 48.5% at single-shot decoding and 89.7% under best-of-4 sampling. Verifiable-reward post-training with GRPO against a 660-entry sequence motif registry improves the useful-plasmid rate across all sampling budgets. We release the 19.3M-parameter model, evaluation suite, and a paired benchmark of prompt-sequence pairs.
|
|
Scooped by
mhryu@live.com
May 21, 11:46 PM
|
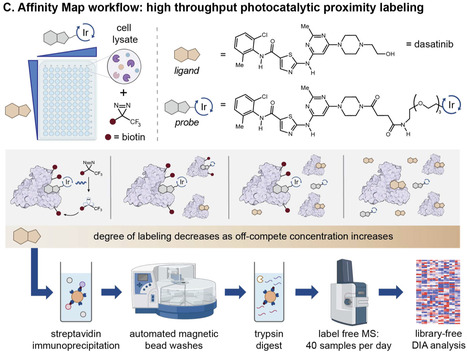
Protein-ligand binding, selectivity, and affinity dictate the effects of drugs and endogenous molecules in cells. Currently, potential protein-ligand interactions are identified by qualitative interpretation of proteomic, transcriptomic, or genomic data, then binding affinities of hits are measured using purified proteins or engineered reporter systems to validate and quantify the strength of individual interactions. Few methods enable simultaneous target identification and biophysical affinity measurement, and these either apply to specific enzyme classes or proteins with ligand-dependent shifts in stability. Here we describe a general platform, termed Affinity Map, which leverages competitive binding analysis, high fidelity photocatalytic labeling, and high throughput proteomics for global quantitative binding affinity profiling. We show that this method is applicable to major classes of ligands, including small molecules, linear peptides, cyclic peptides, and proteins, and can measure affinities between unmodified ligands and proteins in cell lysates, organ extracts, and live cell surfaces. Profiling ligand-protein binding affinities across the proteome remains difficult. Here, the authors report Affinity Map, a photocatalytic labeling platform that accurately measures binding affinities for small molecules, peptides, and proteins in cell lysates, organ extracts, and live cell surfaces.
|
|
Scooped by
mhryu@live.com
May 21, 6:10 PM
|
Accurate gene prediction remains a major bottleneck in fungal genomics, where lineage diversity and alternative splicing challenge existing ab initio methods. Here, we present geneML, a deep learning-based gene prediction tool tailored to fungal genomes. Across nine reference genomes spanning diverse fungal taxa, geneML improved gene-level F1 score from 64.9 to 67.1 compared to BRAKER3 with protein-based hints, driven by substantially higher recall (69.0 vs. 64.1) at equivalent precision. geneML also remains fast, averaging around 6 minutes per genome on a standard 8-core CPU. A key feature of geneML is its ability to predict alternative transcripts. Compared to Fusarium graminearum Iso-Seq control data, it achieves 41.1% transcript recall and 71.1% precision, outperforming AUGUSTUS (33.8% recall, 48.9% precision), one of the few tools that support isoform prediction. The predicted transcript diversity is consistent with experimentally observed fungal alternative splicing patterns. Reannotation of the curated training dataset further suggests improved biological completeness, with geneML recovering 15.3% more genes containing complete PFAM domains than the reference annotation. These results demonstrate that geneML enables faster, more sensitive, and more biologically informative fungal genome annotation. geneML is available as an open-source command-line tool at https://github.com/hexagonbio/geneML.
|
|
Scooped by
mhryu@live.com
May 21, 5:59 PM
|
Rapid quality assessment and multiple assembly comparison are essential steps while assembling new genomes or re-sequencing known ones. Many available tools used for assembly evaluation produce global metrics, representing assembly quality or overall features, most of them working as command line tools that typically act on large data files and produce long detailed result files, where it is not always easy to identify regions of similarity or difference among different chromosome assemblies. ChromoMapperWeb is a new web tool that takes as input nucmer or QUAST output files, quickly identifies similarities and differences between the compared assemblies, and displays them using both table visualizations and pre-arranged or custom graphics. Graphical displays are interactive and allow progressive zoom levels which, in a few steps, move from full genome to very enlarged views, where even small alignment blocks are easily identified. The program, freely accessible through the web server https://chromomapperweb.ceinge.unina.it/, provides an easy-to-use graphical interface, used for experiment planning and interactive evaluation of the results, which include tables and graphical representations of whole genomes, chromosomes, or single blocks.
|
|
Scooped by
mhryu@live.com
May 21, 5:53 PM
|
Microbe Decoder is a web server that predicts functional traits of microbes in microbiome sequencing datasets. Sequencing has revealed thousands of organisms in most ecosystems, but the functional traits of many organisms remain unclear. Existing tools can predict names of organisms or their genes, but they rarely predict concrete biological functions (e.g. fermentation or anaerobic growth). Microbe Decoder fills this gap using three complementary tools relying on taxonomy, metabolic networks, or machine learning. These tools accept either names or gene functions as inputs and are integrated into an easy-to-use web app. When tested against data for microbial isolates, Microbe Decoder showed good predictive performance (e.g. balanced accuracy of 0.85). When applied to datasets from the gut, sediment, and sea, it predicted shifts in functional traits over space and time. Microbe Decoder is designed for use with prokaryotes, with the goal of including eukaryotes in the future. By revealing functional traits of microbes in biological systems, Microbe Decoder will advance biology, medicine, and environmental science. Microbe Decoder is available at https://www.microbe-decoder.org/.
|
|
Scooped by
mhryu@live.com
May 21, 5:43 PM
|
Fungi are a key component of the microbial community in soils, forming species-rich assemblages in soils of natural ecosystems and managed soils. Their unique physiology, absorptive nutritional mode and growth form as spore-producing filamentous eukaryotes enables them to make specific contributions to a wide range of ecological roles as potent decomposers, versatile mutualists and also destructive pathogens. Fungi have important roles in biogeochemical cycles, for example, in nutrient mineralization, plant nutrient uptake and carbon storage. Positioned at the basis of the soil food web, they are one of the pillars of the flow of carbon and energy through the soil food web. As bottom-up controlled organisms in the soil, largely controlled by their resources, they react sensitively to a range of anthropogenic factors, including climate change and other factors of global environmental change, such as chemical pollution. By influencing the food system and via their role as pathogens and in antifungal resistance, fungi are key players in One Health and planetary health. In this Review, Rillig explores the diversity of fungi in the soil ecosystem, their ecological interactions and diverse ecological roles in terrestrial ecosystems as well as anthropogenic factors that affect soil fungi.
|
|
Scooped by
mhryu@live.com
May 21, 5:27 PM
|
The limited availability of structurally specific hydroxylated tryptophan derivatives, whether they are obtained through natural extraction or chemical synthesis, presents challenges for industrial applications. In this study, we report a modular biosynthetic platform in E. coli for producing melatonin and related indoleamines by functionally integrating an animal hydroxylase, an insect decarboxylase, and an avian N-acetyltransferase as well as a plant O-methyltransferase. Following the rational design of key enzymes as well as metabolic pathway optimization, the recombinant strains produced 22.8 g/L of 5-hydroxytryptophan (5-HTP) in a 70 L bioreactor and 6.2 g/L of melatonin in a 5 L bioreactor with exogenous tryptophan feeding. This engineered microbial system establishes an efficient route for manufacturing pharmaceutically relevant tryptophan derivatives and provides a versatile enzymatic toolkit for exploring hydroxylated indole compound biochemistry.
|
|
Scooped by
mhryu@live.com
May 21, 4:46 PM
|
Biomolecular condensates have emerged as versatile regulators of plant cellular processes, offering a dynamic and reversible mechanism to coordinate development, stress response, and spatial organization. Through phase separation, these condensates spatially and temporally modulate biochemical reactions, sequester or activate specific proteins and RNAs, and reshape cellular architecture. This review presents a comprehensive and multidimensional framework for understanding biomolecular condensates in plant biology, from their biophysical properties and ensemble dynamics to their roles across diverse cellular compartments, including plasma membranes, cytoskeleton, intracellular compartments, and chromatin. We highlight their functions in growth, environmental sensing, and defense and discuss current challenges in studying their composition, material properties, and context-dependent behaviors. Understanding plant condensates not only deepens our knowledge of plant cell organization and adaptability but also opens new avenues for biotechnological innovation in agriculture.
|































RNA polymerase activity might be encoded by shorter RNA motifs.