Your new post is loading...

|
Scooped by
mhryu@live.com
Today, 5:15 PM
|
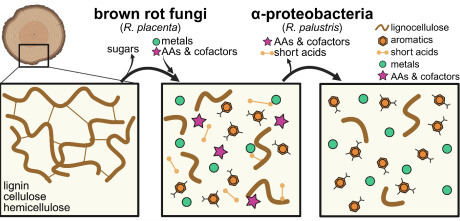
Brown rot wood-degrading fungi release carbon (C) from deadwood but leave behind a large fraction of C sequestered in lignin residues or as fungal metabolites. The strength of sequestration in these C residuals remains unclear, but proteobacteria-dominated bacterial communities have been implicated in metabolizing C from decay residues, possibly erasing the C sequestration potential assumed for brown rot. Here, we paired a model brown rot fungus (Rhodonia placenta) with a model Alphaproteobacterium (Rhodopseudomonas palustris) to track fungal release and bacterial utilization of C derived from decaying wood. We found that fungal decay products generated by R. placenta could be used by R. palustris for growth, and later decay stages contained more usable substrates than early stages. High performance liquid chromatography with mass spectrometry identified a range of aromatic and non-aromatic compounds in the fungal-decayed wood, but after 95 days of bacterial growth, R. palustris preferentially consumed non-aromatic acids over aromatic lignin monomers. Genes involved with aromatic compound degradation were unimportant for bacterial growth, and RNA sequencing revealed that aromatic compound degradation genes were repressed on decayed wood extract. Randomly barcoded transposon sequencing failed to identify a solitary catabolic pathway used by R. palustris, suggestive of substrate co-utilization, and surprisingly, showed that genes involved with copper toxicity were essential. Finally, we found that genes involved with biosynthesis of certain cofactors and amino acids were no longer essential on decayed wood extract, suggesting these nutrients were readily accessible. This study helps lay the foundation to understand potential bacterial-fungal interactions in decayed wood.
|
|
Scooped by
mhryu@live.com
Today, 5:02 PM
|
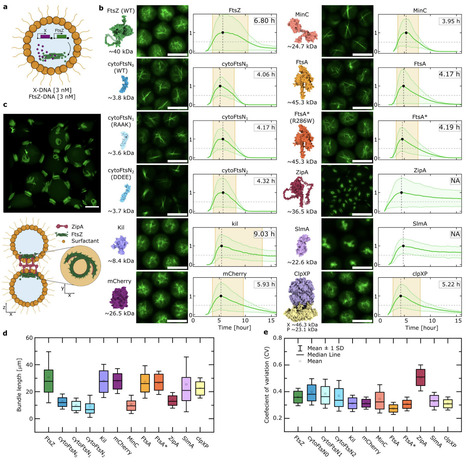
Designing minimal biological systems with emergent functions such as spatiotemporal self-organization is a central goal of bottom-up synthetic biology. While computational optimization and design show promises to accelerate functional protein engineering through Design-Build-Test-Learn cycles, screening libraries for complex functions remains a major challenge. Conventional screens typically lack the spatiotemporal resolution and cell-like confinement required in bottom-up synthetic biology. Here, we present PUREdrop, an automated microfluidic platform that encapsulates and expresses protein libraries in thousands of picolitre-sized synthetic cells per construct. These droplets are sorted into a 96-well plate and analyzed by time-lapse imaging, allowing parallel quantification of expression kinetics and emergent functions. To demonstrate the platform's potential, we first screened computationally re-designed variants of the bacterial cell division protein FtsZ, identifying variants with improved bundling phenotypes and faster kinetics. We then extended our screening procedure towards general protein modulators of FtsZ and identified a combination that anchors filaments to the interface, producing a ring-like phenotype. PUREdrop bridges computational protein engineering and synthetic cell research, elevating the rational engineering of complex biological function to the next level.
|
|
Scooped by
mhryu@live.com
Today, 4:50 PM
|
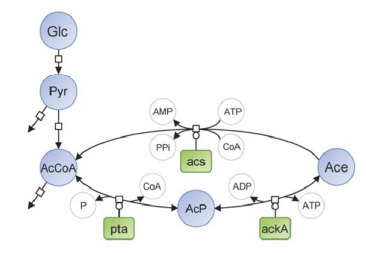
The long-held view that acetate, one of the main fermentation by-products of E. coli, is toxic to microbial growth is currently challenged. Here, we demonstrate that acetate promotes E. coli adaptation to nutrient changes by accelerating growth resumption, with as little as 250 μM acetate being sufficient to shorten the lag phase by several hours. Acetate was found to be consumed via acetyl-CoA synthetase very early after the nutrient change. Transcriptomics, metabolomics and 13C-isotope labeling experiments show that acetate replenishes metabolic pools in the tricarboxylic acid cycle and upper glycolysis. Single-cell analyses reveal that acetate increases the adaptation speed of individual cells switching to the new nutrient. We conclude that the reuse of excreted acetate by E. coli facilitates metabolic adaptation by transiently replenishing central metabolite pools. This work identifies an unexpected role of acetate in the nutritional adaptation of E. coli, providing new insights into the physiological relevance of overflow metabolism.
|
|
Scooped by
mhryu@live.com
Today, 4:15 PM
|
The eukaryotic genome is non-randomly organized within the nucleus, with positioning linked to function. Still, genome-wide radial maps are missing for the majority of experimental model systems. We adapted Genomic loci Positioning by Sequencing (GPSeq) to Saccharomyces cerevisiae, enabling high-resolution mapping along the nuclear cente–periphery axis. GPSeq confirms known spatial features and shows that peripheral telomeres and centromeres impose long-range constraints extending up to 200 kb, restricting short chromosome arms from the nuclear interior. Telomere repositioning to the nuclear center, either artificially or during quiescence, reorganizes much of the genome through inward movement of sub telomeric regions and compensatory shifts of mid-arm chromatin outward. In quiescence, reduced centromere peripheral localization further alters genome organization. While transcription has a modest impact on radial positioning in all studied conditions, we uncover that in the absence of centromere or telomere constraints, GC–content functionally organizes chromatin in the nucleus.
|
|
Scooped by
mhryu@live.com
Today, 3:15 PM
|
With the upgrading of global health demands and safety controversies surrounding artificial sweeteners, natural sweeteners have emerged as a research hotspot thanks to their low-calorie content, high safety, and potential physiological activities. Traditional plant extraction methods are limited by bottlenecks such as long growth cycles and geographical dependence, while synthetic biology has provided an innovative pathway for the efficient and sustainable production of natural sweeteners through metabolic engineering and enzyme engineering, by constructing microbial cell factories. This review systematically summarizes the metabolic pathways and microbial synthesis methods of 12 representative natural sweeteners (including sweet proteins, terpenoid glycosides, flavonoids, polyols, and monosaccharides). By analyzing the advantages and disadvantages of different synthetic schemes as well as the core technical bottlenecks in current production processes, and integrating the latest research progress, this review provides theoretical support and technical references for the future industrial optimization and production of natural sweeteners.
|
|
Scooped by
mhryu@live.com
Today, 1:39 PM
|
Metagenomic sequencing provides insights into microbial communities, but it can be compromised by technical biases, including cross-sample contamination. This phenomenon arises when microbial content is inadvertently exchanged among concurrently processed samples, distorting microbial profiles and compromising the reliability of metagenomic data and downstream analyses. Existing detection methods rely on negative controls, which are insufficiently used and do not detect cross-contamination within non-control samples. Meanwhile, strain-level bioinformatics approaches do not distinguish contamination from natural strain sharing and lack sensitivity. To fill this gap, we introduce CroCoDeEL, a decision-support tool for detecting and quantifying cross-sample contamination. Leveraging linear modeling and a pre-trained supervised model, CroCoDeEL identifies specific contamination patterns in species abundance profiles. It requires no negative controls or prior knowledge of sample processing positions, offering improved accuracy and versatility. Benchmarks across three public datasets demonstrate that CroCoDeEL can detect contaminated samples and identify their contamination sources, even at low rates (<0.1%), provided sufficient sequencing depth. Application of CroCoDeEL to several existing studies reveals previously undetected contamination. Metagenomic analyses of microbial communities can be compromised by technical biases including cross-sample contamination. Here, the authors present CroCoDeEL, a control-free method for detecting cross-sample contamination in metagenomics.
|
|
Scooped by
mhryu@live.com
May 8, 3:00 PM
|
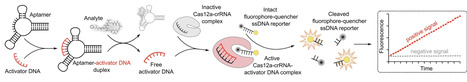
Analyte detection through aptamer-induced signal generation by CRISPR-Cas enzymes has rapidly emerged as a popular biosensing approach. Here, we investigated the implementability and analytical performance of this setup for the detection of diverse small molecule analytes. We selected nine aptamers from the literature targeting seven analytes and tested a commonly used assay design whereby analyte binding by the aptamer liberates a short complementary ssDNA strand, which in turn activates Cas12a to generate a fluorescence signal. After extensive optimization, the assay functioned for only two of the seven analytes, and several previously reported results could not be reproduced. While Cas12a fluorescence ssDNA detection was robust, the low success rate is likely due to aptamers not functioning reliably, underscoring the need for careful aptamer validation. Overall, our study provides a critical assessment of aptamer-Cas12a assay performances and discusses potential strengths, limitations, and pitfalls of this biosensing strategy.
|
|
Scooped by
mhryu@live.com
May 8, 2:01 PM
|
Nisin, a natural, nontoxic antimicrobial peptide, has been widely studied for its broad-spectrum antibacterial activity against Gram-positive bacteria and its applications in food preservation and pharmaceuticals. However, the low nisin titer of Lactococcus lactis poses an obstacle to its large-scale industrial production. Here, we studied the metabolic basis of increased nisin production under aerobic conditions and proposed a high-efficiency fermentation strategy. Aerobic cultivation of L. lactis CF6 significantly increased intracellular ATP and the NAD+/NADH ratio, thus redirecting metabolic flux toward nisin production. Furthermore, cysteine availability and the supplementation of hemin and antioxidants effectively reduced oxidative stress caused by high dissolved oxygen, leading to a significant improvement in nisin titer. Through multimodule synergistic regulation and process optimization, a nisin titer of 18200 IU/mL was achieved in a 5 L bioreactor. In summary, this study lays the foundation for the metabolic engineering of L. lactis and provides valuable insights into the large-scale biomanufacturing of natural antimicrobial peptides.
|
|
Scooped by
mhryu@live.com
May 8, 1:53 PM
|
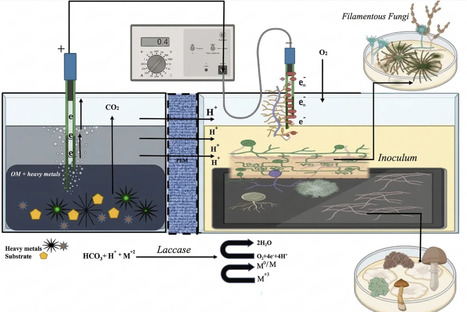
Heavy metal contamination of soil and water is an escalating global environmental issue driven by industrialization and poor waste management. This problem is no longer confined to urban areas, as even small towns are grappling with severe heavy metal contamination, posing substantial threats to human health and aquatic ecosystems. To address these issues, fungal fuel cells have become one of the most promising and environmental friendly technologies. This technology is at the forefront of efforts to combat heavy metal contamination. This method utilizes the unique properties of fungi at the biocathode to treat contaminants and remove heavy metals from soil and water. In addition to reducing pollution, this technology has the capacity to generate electrical current which can serve as an alternative to conventional remediation methods. This review aims to provide a general overview of the fungal fuel cells as a method of bioremediation to remove toxic heavy metals and while simultaneously generating electricity. This review is based on a critical analysis of the recent peer-reviewed publications focusing on the development, operation, and application of fungal fuel cells in heavy metal remediation and bioelectricity production. By exploring the potential of fungal fuel cells, the review provides insight into a future where heavy metal pollution is effectively curtailed while contributing to sustainable energy production, thereby fostering a cleaner and healthier environment.
|
|
Scooped by
mhryu@live.com
May 8, 1:38 PM
|
Developing engineered genetic switches that can respond to specific stimuli in mammalian systems has great potential to advance next-generation therapies. This study introduces a novel tetracycline-inducible artificial riboswitch that regulates gene expression at the translational level via a modified −1 programmed ribosomal frameshifting (−1 PRF) platform. Our dose-response analysis reveals an EC50 of 0.41 μM, indicating exceptional sensitivity compared to other tetracycline-dependent artificial riboswitches developed for mammalian applications. By varying the precise spacing and composition of the tetracycline aptamer as the stimulatory structure on the −1 PRF platform, a series of switches has been constructed that enable robust induction of gene expression upon ligand addition. To attain a translational gene control platform with broader applicability, we developed a concept that allows it to be integrated upstream of a gene of interest (GOI), enabling precise control without requiring extensive re-engineering and expressing the GOI as a fusion protein. We validate the platform’s effectiveness in several genetic contexts, confirming its modularity. As optimization efforts continue to develop artificial riboswitches with optimized properties in mammalian systems, our findings contribute to the development of highly sensitive and efficient regulatory devices with beneficial characteristics, such as reversibility, modularity, and compatibility with RNA-based delivery therapies.
|
|
Scooped by
mhryu@live.com
May 8, 1:30 PM
|
PockFlex is a web server designed to analyze pockets across protein structural ensembles and support the reconstruction, characterization, and prioritization of recurrent binding site organizations. Applicable to ensembles derived from molecular dynamics simulations, multiple experimental structures, or protein structure predictions, PockFlex detects pockets independently in each conformation, retains those overlapping a user-defined region of interest, and groups them across the ensemble by residue-level similarity. This residue-centred clustering framework identifies recurrent binding site clusters, quantifies residue recurrence and variability, and distinguishes persistent from transient binding site regions across the ensemble. Pocket-level druggability, predicted using the PockDrug workflow, is summarized at the cluster level to support binding site prioritisation under conformational variability while preserving access to individual pocket scores. The web application provides interactive, residue-level insights into pocket organisation, variability, and druggability in structural ensembles. The web server is free and open to all users, without login requirement, at https://pockflex.rpbs.univ-paris-diderot.fr/.
|
|
Scooped by
mhryu@live.com
May 8, 9:25 AM
|
A non-fluent English speaker struggles to navigate language barriers in academic publishing. such programmes are rare and don’t always have capacity to meet the demand, notes evolutionary ecologist Andrew McAdam at the University of Colorado Boulder. He helped to create an online platform called Peer Edits that enables authors to solicit feedback on their manuscripts from early-career volunteers working in the same field. Another online resource, Rising Scholars, which provides support for scientists based in low- and middle-income countries, also offers free workshops and mentoring programmes for scientific writing. Laxman Gnawali, the president of the Nepal English Language Teachers’ Association in Kathmandu, adds that he’s found the online university Coursera’s course on academic writing helpful.
|
|
Scooped by
mhryu@live.com
May 8, 1:15 AM
|
Hyperuricemia has emerged as the fourth most prevalent metabolic disorder, necessitating the development of safer and more effective therapeutic strategies. In this study, we constructed a recombinant probiotic strain expressing the PucL and PucM enzymes, which demonstrated a uric acid degradation rate of 65% in vitro. To enhance this activity, we performed modular optimization by employing three ribosome binding sites (RBSs) of different strengths—RBS 29, RBS 31, and RBS T7—to tune the expression levels of pucL and pucM, resulting in highly efficient uric acid degradation. Further improvement was achieved by overexpressing the uric acid transporter gene ygfU and the hydrogen peroxide–degrading catalase gene katG, leading to significant uric acid degradation. Furthermore, the engineered E. coli Nissle 1917 strain was evaluated in a mouse model of hyperuricemia; treatment with the optimized probiotic reduced serum uric acid levels to 39.11 mg/L, representing a 15.98% decrease compared with the control group. Further analysis revealed that this engineered bacterium ameliorates hyperuricemia by modulating the Firmicutes-to-Bacteroidetes ratio, increasing microbial diversity, and promoting the growth of beneficial genera. Collectively, this study establishes an engineered probiotic cell factory for uric acid degradation and demonstrates a proof-of-concept for the microbial remediation of hyperuricemia.
|
|
|
Scooped by
mhryu@live.com
Today, 5:11 PM
|
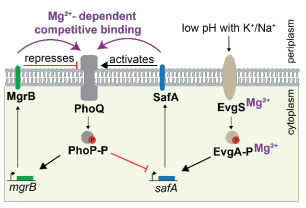
Small proteins are emerging regulators of bacterial virulence via signaling pathways. However, how multiple small proteins coordinate to control a single pathway remains unclear. Here we show that two antagonistic small proteins, SafA and MgrB, forming a magnesium-dependent control module, enhance the signaling sensitivity of the PhoQ/PhoP two-component system in E. coli. Leveraging conditions that enable sequential expression of SafA and MgrB, we dissect and characterize their regulatory dynamics. Our results reveal magnesium as the central control parameter modulating the abundance and affinities of the small proteins for the sensor kinase PhoQ. This coupling together with the competitive binding of the small proteins enables self-adjustment of the signaling network and enhances its sensitivity to environmental magnesium changes. Functionally, loss of either small proteins influences the ability of enteropathogenic E. coli to evade macrophage phagocytosis, linking this regulatory scheme to host interaction. Our findings establish a framework for achieving a high level of input sensitivity through antagonistic regulations in a signaling network.
|
|
Scooped by
mhryu@live.com
Today, 4:56 PM
|
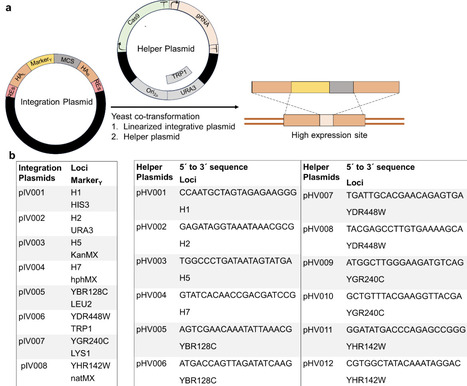
Budding yeast Saccharomyces cerevisiae is a workhorse chassis for producing added food and agricultural compounds. However, building multi-enzymatic pathways for these chemicals often requires iterative genomic integration, underscoring the need for efficient, rapid genome-editing tools that can reliably target transcriptionally active chromosomal regions. In this study, to accelerate strain construction, we established a genome-editing toolkit to rapidly engineer eight loci, highly expressed hot-spots, but nonessential genomic sites suitable for stable pathway assembly. Our approach integrates three key design features: (i) selectable markers to enable rapid screening of edited cells, (ii) extended homology arms that leverage the yeast homology-directed repair machinery for robust genomic integration, and (iii) co-delivery of Cas9 and guide RNAs to promote efficient double-stranded DNA breaks at specific integration sites. The sequence independence of FASTOP relies on the release of integration cassettes from integrative vectors, mediated by restriction digestion at two flanking multiple-cutting sites in the integration module to minimize the risk of introducing sequence errors during PCR amplification of the integration cassettes. Following the introduction of a fluorescent reporter cassette, we observed high integration efficiencies across the target sites. We then integrated the biosynthetic pathway of plant-derived flavonoid naringenin into the hot-spots of the yeast genome using the FASTOP toolkit. Our results demonstrated that upon expressing the five essential genes in simple shake flask culture, naringenin production reached 505.7 mg/L, representing a significant (69-fold) increase over previously reported titers for comparable minimal heterologous pathways in S. cerevisiae. Together, the FATSOP toolkit provides a user-friendly platform for reliably modifying hot-spot loci to rapidly construct multi-enzymatic metabolic pathways in S. cerevisiae, while achieving high production levels for high-value food-relevant metabolites.
|
|
Scooped by
mhryu@live.com
Today, 4:35 PM
|
Crop nutrition depends on plant-microbe interactions, yet it remains unclear whether conserved genetic pathways impose universal rules on root microbiome assembly across plant hosts. Here, we show that the Common Symbiosis Signalling Pathway (CSSP), a conserved genetic module controlling endosymbiosis with arbuscular mycorrhizal fungi and nitrogen-fixing bacteria, regulates root microbiome assembly in a host-specific manner across contrasting fertilization regimes. Using Lotus japonicus and Hordeum vulgare, we demonstrate that mutations in orthologous CSSP genes remodel root bacterial communities in both species, but with distinct taxonomic outcomes. In Lotus, CSSP disruption reduces rhizobial colonization and promotes niche replacement by commensal taxa, whereas in Hordeum, the same mutations broadly restructure bacterial lineages without converging on Lotus-like responses. Root exudate profiling reveals host-specific metabolic differences, particularly in phenylpropanoid (flavonoids and coumarins) and gibberellin pathways, linking CSSP activity to chemically distinct rhizosphere environments that correlate with divergent microbiome assembly patterns across hosts. Moreover, root bacterial community composition accurately predicts plant nutritional status, highlighting tight coupling between host physiology and microbiome composition. Together, our results show that conserved symbiosis signalling regulates root microbiome assembly, while host-specific metabolic environments determine taxonomic outcomes. This extends CSSP function beyond canonical endosymbioses and positions symbiosis signalling as a general determinant of plant-microbiome interactions with implications for crop nutrition.
|
|
Scooped by
mhryu@live.com
Today, 3:23 PM
|
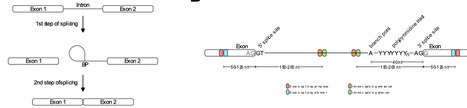
Variants affecting RNA splicing are a major contributor to human disease, yet the consequences of variants outside of the canonical splice motifs are often difficult to determine. Here, we present a protocol for minigene-based evaluation of candidate splice-altering variants. The methodology described includes locus-specific insert design, commercial gene fragment synthesis, and long-read sequencing. The combined approach enables rapid assay development and nucleotide level resolution of the effect on splice isoforms in vitro, providing a scalable framework for functional validation of predicted cryptic splice variants.
|
|
Scooped by
mhryu@live.com
Today, 3:11 PM
|
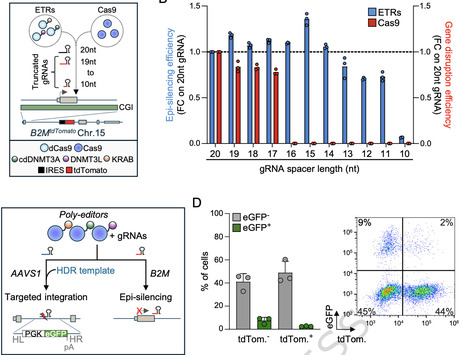
The parallel disruption of multiple genes coupled with targeted transgene insertion offers a powerful strategy for more effective and precise cell engineering. However, such orthogonal editing involves the induction of multiple DNA breaks, raising safety concerns related to the risks of chromosomal translocations. Here, we present a polyfunctional CRISPR-Cas9-based strategy that enables both transgene insertion and epigenetic silencing at distinct genomic loci in a single treatment without inducing reciprocal chromosomal translocations. This is accomplished through an optimized all-in-one epigenome editor equipped with a catalytically active Cas9, whose endonuclease activity is selectively disabled at epigenetically silenced loci using truncated gRNAs. As a proof of concept, we demonstrate that this platform enables efficient multi-locus editing, including functional replacement of the endogenous TCR with a tumor-selective one, targeted insertion of a prototypic CAR with either a selectable marker or an immunomodulatory receptor into a TCR locus or a ubiquitously expressed gene, and durable, multiplexed epigenetic silencing of clinically relevant genes in primary human T cells. Polyfunctional editing establishes a versatile and safe framework for orthogonal editing, broadening the scope of genome and epigenome engineering in cancer immunotherapy and beyond. Cell engineering through multiple genetic edits holds therapeutic potential but raises safety concerns. Here, authors develop a CRISPR-Cas9 molecule enabling simultaneous transgene insertion and multi-gene epigenetic silencing, proving efficacy and safety in human T cells for cancer immunotherapy.
|
|
Scooped by
mhryu@live.com
May 8, 3:02 PM
|
Long before nature was ‘red in tooth and claw’, it was sundered by nano-spears and seeping poisons. Microorganisms were the first predators, and predation has since deeply shaped all major branches of life — from individual traits to collective systems, community dynamics and major evolutionary transitions. Yet, we have only begun to understand how microbial predation influences the genetics, morphology, behavior, ecology and evolution of microorganisms in natural communities and, in turn, the macroscopic biosphere. With the field advancing rapidly on diverse fronts, integrative conceptual frameworks, questions and research approaches are needed to promote synthetic development of the field. In this Review, we explore the remarkably diverse forms of microbial predation that have evolved so far, considering organismal traits and their molecular foundations alongside the evolutionary ecology of predator–prey interactions in community contexts. Building on a process-based definition, forms of microbial predation are conceptualized along gradients, including gradients of evolutionary adaptedness for predation and of privatization of prey-derived nutrients. Important future research themes include predation origins and early stages of predatory adaptation, effects of diverse forms of predation on community diversity and stability, predator–prey co-evolution in complex communities, and multi-approach development of unicellular predators as biocontrol agents. In this Review, Vasse and Velicer explore the phylogenetic and functional diversity of predators of microorganisms, conceptualizing the forms of microbial predation along gradients, including gradients of evolutionary adaptedness for predation and privatization of prey-derived nutrients.
|
|
Scooped by
mhryu@live.com
May 8, 2:48 PM
|
The probiotics field, a historically popular yet scientifically debated discipline, is moving beyond a decades-long promotion of ‘first-generation’ food-derived strains towards the development of ‘next-generation probiotics’ (NGP) or ‘precision probiotics’, natural and engineered strains featuring improved human colonization, clinical efficacy and safety profiles. In this Review, we outline the evolution of NGP and means by which their development is designed to tackle challenges of live bacterial therapy related to colonization resistance, in-host evolution, long-term safety and insufficient understanding of therapeutic and off-target mechanisms of activity. We showcase how a variety of emerging strategies enable the identification of NGP strains and define consortia featuring therapeutic potentials in metabolic, immune and oncological diseases. Finally, we discuss how computational and artificial intelligence (AI) advances can reshape NGP development, including AI-based discovery of strains and bioactive compounds; computational-driven design of engineered microorganisms and multi-kingdom consortia; and AI-assisted structural and metabolic network-based modelling predicting personalized NGP function, interactions and therapeutic impacts. In this Review, Kern, Tofield, Frame and Elinav discuss recent advances in the design of next-generation probiotics, from identification of candidates to therapeutic applications across diverse disease contexts, and highlight major challenges and the potential of artificial intelligence to develop effective, personalized probiotics with therapeutic functions.
|
|
Scooped by
mhryu@live.com
May 8, 1:56 PM
|
Chitin and its deacetylated derivative, chitosan, are natural polymeric polysaccharides derived from crustaceans, fungi, and insects with antimicrobial, anti-inflammatory, antioxidant, and biocompatible bioactivities. As a complement to crustacean biomass, fungi provide a sustainable source of chitin/chitosan. Recent biotechnology and materials science advancements have stimulated significant research interest in applying fungal-derived chitin and chitosan for wound healing materials. This paper comprehensively reviews fungal chitin/chitosan-based materials for wound healing applications, encompassing innovative forms, such as nanoparticles, membranes, hydrogels, sponges, microfibrous nonwovens, and nanofibrous scaffolds. Moreover, this Review elucidates the limitations of using fungal chitin and chitosan in wound healing applications, including low purity and extraction yield, inadequate quality control measures, suboptimal biocompatibility, and insufficient evaluation of action mechanisms. To advance the innovation and commercialization of fungal-derived chitin- and chitosan-based wound healing materials, it is imperative to enhance multidisciplinary collaboration among microbiology, chemistry, materials science, and clinical medicine.
|
|
Scooped by
mhryu@live.com
May 8, 1:50 PM
|
The accelerating global crisis of antimicrobial resistance (AMR) demands rapid and accurate methods for antibiotic susceptibility testing (AST). Conventional phenotypic assays remain the gold standard but are hindered by long culture times, while genotypic tests cannot reliably predict phenotypic resistance. In recent years, metabolism-based AST has emerged as a promising alternative, enabling the rapid detection of bacterial responses to antibiotics through shifts in metabolic activity. These approaches bridge molecular speed with phenotypic precision, allowing susceptibility determination within hours, or even minutes, without requiring cell proliferation. In this review, we summarize the latest advances in metabolism-based biomarkers for rapid AST. First, we discuss how antibiotics influence bacterial metabolism, linking resistance mechanisms to metabolic activities. We then summarize emerging metabolic biomarkers, categorized by their physiological underpinnings: nutrient uptake, respiratory activity, metabolic reprogramming, and enzymatic function. Finally, we list key challenges and future directions toward deployable metabolism-based AST platforms.
|
|
Scooped by
mhryu@live.com
May 8, 1:33 PM
|
mRNA-based therapeutics have revolutionized the treatment and prevention of infectious, neurological, and cancer diseases. However, their linear topology makes them susceptible to rapid degradation in vivo, which limits their therapeutic efficacy. Engineered circular RNAs (circRNAs) due to their closed ends and high stability are emerging as a promising alternative to linear RNA therapies. Engineered circRNAs are also increasingly used to mimic naturally occurring circRNAs in functional studies. Both applications, however, depend on production of precise circRNAs with homogenous sequences to enable accurate interpretation of biological outcomes. To address this, we developed and optimized methods for generating precise circRNAs. We employed enzymatic ligation of linear RNAs rather than autocatalytic splicing to produce circRNAs to minimize extraneous nucleotides remaining from the ribozymes. We carefully designed the DNA transcription template to maintain sequence and structural integrity. A permuted DNA template leveraging three internal guanosines (Gs) was synthesized and amplified using a reverse primer containing two 2′-O-methyl groups. This approach optimally produced the linear precursor RNA with correct 5′ and 3′ ends. After testing multiple workflows, we found that GMP-primed in vitro transcription, T4 RNA ligase 2-mediated circularization, and urea–polyacrylamide gel electrophoresis (PAGE) gel extraction produced the highest fidelity circRNAs.
|
|
Scooped by
mhryu@live.com
May 8, 11:20 AM
|
Natural products, as metabolites from microorganisms, animals or plants, exhibit diverse biological activities, making them crucial for drug discovery. Nowadays, existing deep-learning methods for natural products research primarily rely on supervised learning approaches designed for specific downstream tasks. However, such one-model-for-a-task paradigm often lacks generalizability and leaves substantial room for performance improvement. Additionally, existing molecular characterization methods are not well-suited for the unique tasks associated with natural products. Here, to address these limitations, we pretrained a foundation model for natural products (NaFM) based on their unique properties. Our approach employs a pretraining strategy specifically tailored to natural products. By incorporating contrastive learning and masked graph learning objectives, we emphasize evolutional information from molecular scaffolds while capturing side-chain information. NaFM achieves state-of-the-art results in various downstream tasks related to natural product mining and drug discovery. We first compare taxonomy classification with synthetic molecule-focused baselines to demonstrate that current models are inadequate for understanding natural synthesis. Furthermore, by diving into a fine-grained analysis at both the gene and microbial levels, NaFM demonstrates the ability to capture evolutionary information. Eventually, our method is applied to virtual screening, illustrating informative natural product representations that can lead to more effective identification of potential drug candidates. Ding et al. present a scaffold-aware foundation model for small-molecule natural products leveraging masked objectives and contrastive learning to enhance taxonomy classification, genome mining and virtual screening in drug discovery.
|
|
Scooped by
mhryu@live.com
May 8, 9:20 AM
|
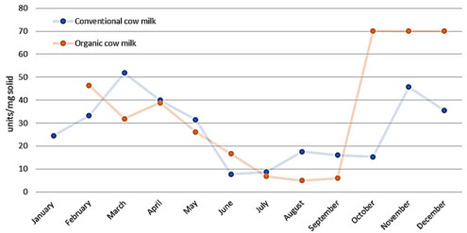
Milk is one of the most vital foods worldwide, valued not only for its nutrient-rich composition but also for its diverse range of bioactive compounds. In addition to its nutritional importance, milk contains a variety of proteins that serve significant biological functions. Among these are lactic acid bacteria (LABs), which can produce antimicrobial peptides and organic acids. The antimicrobial effects of milk are primarily attributed to its bioactive proteins, including whey proteins and caseins. Whey proteins, such as lactoferrin, lysozyme, and immunoglobulins, as well as peptides derived from these proteins, exhibit significant antimicrobial activity, particularly against Gram-positive and Gram-negative bacteria. These peptides are released during proteolysis, either through enzymatic digestion or fermentation, and can interact with bacterial membranes, destabilising them and preventing microbial growth. The concentration of antimicrobial proteins varies across mammalian milks, with higher levels often observed in species such as sheep and goats, reflecting adaptations to specific environmental and immune challenges. Despite the reduction in antimicrobial efficacy following heat treatments like ultra-high temperature (UHT) or pasteurisation, fermented dairy products such as yoghurt and cheese retain significant antimicrobial properties, mainly due to the presence of bioactive peptides and increased acidity. These antimicrobial activities underscore the potential of milk-derived compounds as natural alternatives to antibiotics, particularly in food safety and therapeutic applications. Further research into milk’s bioactive peptides could expand their use in the prevention and treatment of microbial infections.
|