 Your new post is loading...

|
Scooped by
mhryu@live.com
April 3, 5:21 PM
|
Soil microbial ecosystems are complex and difficult to replicate in laboratory settings. It is often unclear which pressures most strongly shape microbial survival and evolution in situ, and new methods are needed to intersect the manipulative power of the lab with the reality of field environments. One recent innovation was the “isolation chip,” in which many new microbial isolates could be cultured on agar within a buried diffusion chamber while exposed to environmental inputs through fine-pored membranes. Here, we created a modified version of this device containing biologically-cleared soil instead of agar, to trial an in situ reverse ecology experimental evolution approach. Using these “adaptation chips (aChips)” we exposed populations of two different soil-dwelling bacteria (Priestia megaterium and Streptomyces lydicus) to several farm soils in the Northeast US for up to two years, documenting mutations arising in the evolving populations. While evolution was remarkably slow in the field, P. megaterium populations accumulated many mutations pre-burial during aChip construction which seemingly reflected zinc limitation in the aChip carrier soil. Although post-burial mutations were observed in both P. megaterium and S. lydicus populations, they remained at low frequency and did not display parallelism between aChips buried at the same sites, indicating a lack of strong positive selection and/or limited generations of population growth within the aChip. We suggest several improvements to aChip design to facilitate greater evolutionary progression, including a larger within-aChip soil volume and fewer cells initially secured inside the aChip.
|
|
Scooped by
mhryu@live.com
April 3, 5:14 PM
|
Magnetotactic bacteria (MTB) are a group of gram-negative species that produce a lipid-bounded organelle, the magnetosome, in which a magnetic crystal is biomineralized. MTB use magnetosomes to align with the geomagnetic field for improved navigation of their environment. To optimize this alignment, these species linearly organize their magnetosomes using a handful of factors, including the bacterial actin-like protein MamK. Despite these shared features, there is a broad diversity of species-specific linear magnetosome chain arrangements within MTB, but the molecular mechanisms behind these phenotypic variations are unclear. Recently, genetic analyses showed that two proteins—McaA and McaB—interface with the chain organization machinery of Magnetospirillum magneticum AMB-1 to arrange magnetite crystals in a series of subchains rather than the single cohesive chains found in closely related MTB. Here, we use in vivo co-immunoprecipitation in AMB-1 to demonstrate protein-protein interactions between McaA, McaB, and MamK. Experiments with McaA truncation mutants and conditional control of McaB localization determined that McaA-McaB interactions are dependent on amino acids 530–665 of McaA and McaB localization to the magnetosome chain. We further show that disrupting the McaA-McaB interaction alters the spatial dynamics of MamK in vivo. We present a model in which protein-protein interactions between McaA, McaB, and MamK drive changes in MamK behavior to establish AMB-1’s magnetosome chain organization.
|
|
Scooped by
mhryu@live.com
April 3, 5:06 PM
|
The ongoing census of microbial life is hampered by disparate sampling across Earth’s habitats, challenges in isolating uncultivated organisms, limited resolution in taxonomic marker gene amplicons and incomplete recovery of metagenome-assembled genomes. Here we quantify discoverable Bacterial and Archaeal diversity in a comprehensive, curated cross-habitat dataset of 92,187 publicly available metagenomes. Clustering 502 million sequences of 130 marker genes, we predict ~705,000 Bacterial and ~27,000 Archaeal species-level clades, the vast majority of which were hidden among unbinned contigs. We estimate that ten and 145 previously undescribed Archaeal and Bacterial phyla, respectively, are discoverable in this dataset. We identify soils and aquatic environments as hotspots of discoverable lineages, but predict that undescribed taxa remain abundant across all habitats. Finally, we show that prokaryotic diversity appears to arise within common evolutionary patterns, as clade size distributions follow power laws, consistently across the Tree of Life. Re-analysis of over 92,000 metagenomes reveals hundreds of thousands of previously undescribed Bacterial and Archaeal clades hidden in plain sight.
|
|
Scooped by
mhryu@live.com
April 3, 4:55 PM
|
Prokaryotic Argonaute proteins (pAgos) are programmable nucleases that always utilize DNA guides to cleave DNA targets. Recent studies show that some pAgos preferentially utilize DNA guides to cleave RNA targets rather than DNA targets. VbAgo, derived from a Verrucomicrobia bacterium, is a nuclease capable of specifically cleaving single-stranded RNA and highly structured RNA substrates at 37 °C, making it an ideal candidate for developing RNA manipulation toolkits. An in-depth investigation of its mechanism contributes to understanding the functional characteristics of gDtR-type Ago proteins. Here, we present cryo-electron microscopy structures of VbAgo, the VbAgo-guide DNA binary complex, multiple wild-type VbAgo-guide DNA-target RNA ternary complexes, and the catalytically inactive mutant (VbAgo-DM) guide DNA-target RNA ternary complex, with resolutions ranging from 2.5 to 3.2 Å. By integrating these cryo-EM structures with biochemical data, we elucidate the entire catalytic process of VbAgo, revealing its unique C-terminal regulatory mechanism. Specifically, in its apo state, VbAgo’s C-terminus occupies the nucleic acid binding channel, partially impeding its catalytic activity while enhancing its stability. The binding of guide DNA displaces the C-terminus, and subsequent binding of target RNA, along with conformational changes in the N-terminal and PAZ domains, facilitates VbAgo dimerization. Following this, the C-terminus transitions from a loop to a helix, enabling maturation of the catalytic center and inducing movements in the MID-PIWI’ interactions at the dimer interfaces, ultimately leading to dimer dissociation. Concurrently, cleavage of the target RNA and subsequent product release occur, after which VbAgo reverts to its binary state to initiate the next cleavage cycle. Moreover, we demonstrate that VbAgo exhibits guide DNA mediated RNA knockdown activity in mammalian cells. In summary, our study provides a comprehensive understanding of the molecular mechanisms governing self-inhibition, guide binding, target recognition, and product release in VbAgo. These findings offer valuable insights into the diverse mechanisms of pAgos, broadening their functional scope and enhancing the biotechnological potential of pAgo proteins. Prokaryotic Argonaute proteins are programmable nucleases. Here, the authors capturing structures of VbAgo at functional stages to show how its C terminus acts as a regulator and demonstrate its ability to cleave RNA in mammalian cells.
|
|
Scooped by
mhryu@live.com
April 3, 4:48 PM
|
As the generation of data in the life and health sciences expands rapidly, there is a growing need for professionals and students in these fields to master core bioinformatics skills, particularly those relating to Unix-like systems, most commonly used in bioinformatics. This paper introduces two key contributions to address this need: (1) A Unix curriculum for life scientists with little or no command-line experience, based on progressive Unix skill levels for bioinformatics and (2) An implementation of this curriculum into a series of interactive online tutorials deployed through Sandbox.bio—an open-source platform for learning bioinformatics that embeds a command line in the browser, which removes barriers related to software installation and access. We performed an overall evaluation of this teaching framework in different contexts. This inclusive, sustainable approach provides widespread access to essential bioinformatics skills for life science students and professionals alike.
|
|
Scooped by
mhryu@live.com
April 3, 4:30 PM
|
Microorganism-based therapies, particularly those utilizing probiotics, have emerged as a powerful biomedical strategy owing to their inherent living functionalities. These living systems can dynamically interact with host environments and self-regulate their activity, offering superior adaptability, prolonged functionality, and microenvironmental responsiveness compared to conventional non-living therapeutic platforms. Despite these advantages, the direct administration of probiotics faces several challenges, such as poor viability, limited retention at target sites, and the inability to control therapeutic effects in a spatiotemporally precise manner. To address these challenges, embedding probiotics within hydrogel matrices has proven effective in enhancing microbial stability, prolonging in vivo retention, and enabling precise and sustained therapeutic delivery through synergistic interactions between the hydrogels and living microorganisms. This review provides a comprehensive overview of the materials and design strategies employed in the construction of living microorganism-encapsulated hydrogels (living hydrogels), with particular emphasis on the dynamic interactions and synergistic mechanisms of hydrogel-microorganism systems. We further illustrate how these mechanisms can achieve various biomedical applications, such as modulating gut microbiota to treat gastrointestinal disease and accelerate wound healing, or leveraging microbial-induced immune regulation for effective cancer therapy. Finally, the current challenges and future directions associated with the clinical translation of living hydrogels are highlighted. Therefore, the unique multifunctionality and therapeutic promise of living hydrogels position them as compelling candidates for the development of next-generation biomaterials with unprecedented therapeutic potential.
|
|
Scooped by
mhryu@live.com
April 3, 1:46 AM
|
Systems engineering has transformed chemical manufacturing, but bioprocessing has lagged in adopting comprehensive approaches. This review explores strategies that successfully engineer integrated upstream and downstream bioprocesses. Our analysis reveals a critical gap: bioprocess subsystems are typically optimized in isolation (‘subsystems optimization’), which limits the overall performance. We identify four key leverage points for systems engineering: engineering product accessibility to eliminate cell lysis, modifying strains to remove contaminants, adapting products for simplified purification, and enhancing strain tolerance for improved separation. While these integrative approaches substantially improve process consolidation, our findings show that there remains a significant misalignment between academic research and industrial needs (failing commercially relevant metrics). Embracing a holistic systems perspective is essential for future bioprocesses to have a transformative impact.
|
|
Scooped by
mhryu@live.com
April 3, 1:38 AM
|
Sequence-based deep learning models have become the state of the art for analyzing the genomic regulatory code. Particularly for enhancers, these models excel at deciphering sequence grammar that underlies their activity. To enable end-to-end enhancer modeling and design, we developed a software package called CREsted (cis-regulatory element sequence training, explanation and design). It combines preprocessing and analysis of single-cell assay for transposase-accessible chromatin using sequencing data, modeling chromatin accessibility from sequence, sequence design and downstream analysis to decipher enhancer grammar. We demonstrate CREsted’s functionality on a mouse cortex and a human peripheral blood mononuclear cell dataset. Additionally, we use CREsted to compare mesenchymal-like cancer cell states between tumor types, and we investigate different fine-tuning strategies of genomic foundation models within CREsted. Finally, we train a model on a zebrafish development atlas and use this to design and in vivo validate cell-type-specific enhancers. For varying datasets, we demonstrate that CREsted facilitates efficient training and analyses, enabling scrutinization of the enhancer logic and design of synthetic enhancers across tissues and species. CREsted is an efficient and user-friendly toolbox for analysis, modeling and design of cell-type-specific enhancers across diverse species.
|
|
Scooped by
mhryu@live.com
April 3, 12:47 AM
|
Predicting which receptor a phage binds to from genome sequence alone has remained an intractable challenge, principally because the experimental phenotypic data required to train and validate predictive models have not been available at sufficient scale. Here we address this by conducting 1,050 genome-wide genetic screens across 255 taxonomically diverse Escherichia coli dsDNA phages, assigning host receptors to 193 phages across 19 receptor classes. Comparative genomics and AlphaFold3 structural modelling resolved the sequence determinants of specificity to defined receptor-binding protein domains and individual residues. Machine learning models trained on this dataset predicted host receptor identity from phage genome sequence alone without prior annotation of receptor-binding genes, achieving perfect precision and greater than 80% recall on 49 independently validated phages, and yielding predictions for 1,050 of 1,875 E. coli phage genomes in NCBI. Domain swaps redirected receptor specificity as predicted, and a single amino acid substitution proved both necessary and sufficient to switch recognition between two distinct porins. These results demonstrate that systematic phenotyping at scale makes sequence-based prediction of molecular interaction specificity tractable, with direct implications for phage-based medicine, microbiome engineering and the broader challenge of inferring host-pathogen interaction outcomes from sequence.
|
|
Scooped by
mhryu@live.com
April 3, 12:29 AM
|
two research teams describe the machine-learning algorithms they developed to screen bacterial genomes and identify proteins that are involved in protecting the microorganisms against viral invaders. Their analyses identified hundreds of thousands of potential antiviral proteins, which researchers could harness to develop innovative biotechnologies. Laub and his colleagues have made DefensePredictor freely available online for researchers to use. Bernheim and her colleagues have also created an open-access database called DefenseFinder, which contains more than 44,000 predicted antiviral systems. Researchers can use these resources to test the antiviral properties of newly identified proteins.
|
|
Scooped by
mhryu@live.com
April 3, 12:22 AM
|
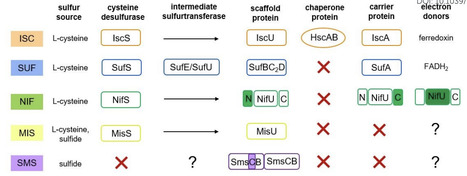
Iron-sulfur (Fe-S) clusters are ancient inorganic cofactors ubiquitous across all domains of life. These cofactors associate with proteins through constitutive or transient coordination, expanding their chemistries and versatility in biological processes. Thus, Fe-S proteins participate in intricate and multifaceted chemistries critical to life on Earth. The biosynthesis of these cofactors has evolved to require complex machinery to catalyze cluster formation and subsequent transfer to target apo-proteins. Five Fe-S cluster biogenesis systems have been identified, with varying degrees of complexity, including: iron-sulfur cluster (ISC), nitrogen fixation (NIF), sulfur mobilization (SUF), minimal iron-sulfur system (MIS), and SUF-like minimal system (SMS). Sulfur mobilization in the biosynthesis of Fe-S clusters is initiated, in most cases, by cysteine sulfurtransferases, also known as cysteine desulfurases. These enzymes use the amino acid cysteine as a sulfur donor and require specific interactions with a sulfur acceptor to promote sulfur transfer. Physical interactions and coordination among biosynthetic components restrict their functions and guarantee the trafficking of reactive intermediates to proper destinations. As recently reported, the occurrence of alternate biosynthetic schemes using sulfide as the sulfur source bypasses the requirement for sulfurtransferases and provides alternate evolutionary strategies to construct Fe-S clusters.
|
|
Scooped by
mhryu@live.com
April 3, 12:01 AM
|
NUPACK is a growing software suite for the analysis and design of nucleic acid structures, devices, and systems serving the needs of researchers in the fields of nucleic acid nanotechnology, molecular programming, synthetic biology, and across the life sciences. NUPACK algorithms have pioneered the treatment of complex and test tube ensembles containing arbitrary numbers of interacting strand species, providing crucial tools for capturing concentration effects essential to analyzing and designing the intermolecular interactions that are a hallmark of these fields. Analysis and design of multitube ensembles enable reaction pathway engineering of dynamic hybridization cascades and structural engineering including the possibility of pseudoknots. The all-new NUPACK 4 scientific code base offers enhanced physical models (coaxial and dangle stacking subensembles), dramatic speedups (20–120× for test tube analysis), increased scalability for large complexes (e.g., 30,000 nt), mixed materials (specified at nucleotide resolution), and diverse hard and soft sequence constraints for design. The all-new NUPACK web app (nupack.org) facilitates rapid job submission and result inspection with NUPACK 4 algorithms running in parallel on a hybrid cloud compute cluster that scales dynamically in response to user demand. NUPACK 4 algorithms can also be run locally using the all-new NUPACK Python module.
|
|
Scooped by
mhryu@live.com
April 2, 11:48 PM
|
Type IV secretion systems (T4SS) are versatile machines with variable functions including DNA uptake and release, protein translocation, and DNA conjugation. However, the diversity, distribution, and functional roles of the T4SS in the Ralstonia genus remain poorly understood. The Ralstonia solanacearum species complex (RSSC) comprises three species of plant-pathogenic bacteria that cause bacterial wilt disease. The Ralstonia genus also includes non-RSSC species that are primarily environmental bacteria and rare opportunistic human pathogens. This study compared the diversity and phylogenetic distribution of T4SSs in the RSSC phytopathogens vs. non-RSSC environmentals. Phylogenetic analysis of VirB4 sequences and synteny analysis revealed 16 distinct T4SS clusters in Ralstonia, with 10 clusters found in RSSC phytopathogen genomes, 12 in non-RSSC environmental genomes, and 6 clusters in both groups. Collectively, these gene clusters were more prevalent in non-RSSC environmental genomes. The presence of type IV coupling protein and relaxase genes suggests that at least 14 of these T4SS gene clusters are putative DNA-conjugation systems. The clusters were encoded on accessory plasmids of various sizes or as integrative and conjugative elements on the chromosome or megaplasmid. The putative regions of transfer for T4SS gene clusters in the RSSC phytopathogen genomes often contained type III effectors, type VI secretion toxin/antitoxin clusters, and haemagglutinin gene clusters. In contrast, the non-RSSC environmentals were enriched in heavy metal metabolism and resistance genes. One of the 16 T4SS clusters, cluster i, exhibited evidence of specialization for the RSSC phytopathogens. These findings shed light on the eco-evolutionary differences within the genus Ralstonia.
|
|
|
Scooped by
mhryu@live.com
April 3, 5:17 PM
|
Aromatic amino acids—tryptophan, tyrosine, phenylalanine, and histidine—are essential for bacterial growth and are among the most energetically expensive metabolites to synthesize. Despite this cost, it has been recently shown that bacteria possess exporters for these amino acids. Here, we identify aexB (formerly yvjA) as a gene encoding a novel aromatic amino acid exporter in Bacillus subtilis. Using a transposon-based screen, we found that aexB overexpression confers resistance to the toxic tryptophan analog 5-fluorotryptophan. Additional analog screens revealed that AexB also promotes tolerance to toxic derivatives of tyrosine, phenylalanine, and histidine but not non-aromatic amino acids. LC-MS analysis showed that AexB specifically exports aromatic amino acids, and co-culture assays confirmed that overexpression of aexB can support the growth of aromatic amino acid auxotrophs. Furthermore, overexpression of aexB impaired growth when intracellular tryptophan was limiting. On the other hand, deletion of aexB exacerbated growth defects under excess tryptophan conditions, likely due to feedback inhibition of aromatic amino acid synthesis pathways. Our findings reveal that AexB is an aromatic amino acid exporter that functions as a metabolic safety valve.
|
|
Scooped by
mhryu@live.com
April 3, 5:10 PM
|
Adenosine-to-inosine (A-to-I) mRNA editing alters genetic information post-transcriptionally and can impact protein sequence and function, yet its regulation in bacteria remains unclear. Here, we profiled A-to-I editing in E. coli across nutrient-rich Luria-Bertani (LB) and minimal M9 media and different growth phases. Our analysis expanded the repertoire of TadA-dependent A-to-I edited mRNAs to 27, including 12 novel sites, and revealed that editing levels were dynamic and markedly increased at the stationary phase in LB but not in M9. Editing levels were independent of mRNA expression yet correlated with tRNA-Arg2 downregulation, and overexpressing tRNA-Arg2 reduced mRNA editing, demonstrating substrate competition for TadA, the sole bacterial tRNA adenosine deaminase. Mutants with TadA-deficient editing or reduced tRNA-Arg2 expression displayed similar LB-specific growth defects. Moreover, tRNA-Arg2 expression, tRNA-Arg2-dependent codon usage, and tRNA-Arg2 editing were all elevated in LB compared to M9. These findings establish regulatory principles for bacterial RNA editing, implicate tRNA editing in nutrient-responsive fitness, and provide a framework to explore the physiological roles of mRNA editing.
|
|
Scooped by
mhryu@live.com
April 3, 4:57 PM
|
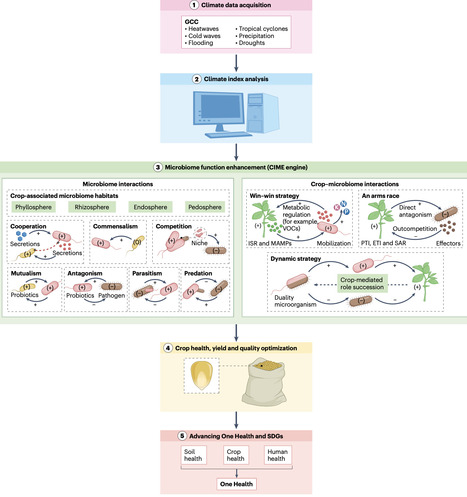
Crop productivity under climate stress remains constrained by conventional agricultural approaches that underuse plant–microbiome interactions. A microbiome-centred, climate-responsive framework was proposed to enhance crop resilience and agro-sustainability by prioritizing targeted manipulation of crop-associated microbiomes, offering a scalable and adaptive pathway to buffer climate stresses and stabilize crop performance.
|
|
Scooped by
mhryu@live.com
April 3, 4:52 PM
|
Plant phenotyping increasingly relies on (semi-)automated image-based analysis workflows to improve its accuracy and scalability. However, many existing solutions remain overly complex, difficult to reimplement and maintain, and pose high barriers for users without substantial computational expertise. To address these challenges, we introduce PhenoAssistant: a pioneering AI-driven system that streamlines plant phenotyping via intuitive natural language interaction. PhenoAssistant leverages a large language model to orchestrate a curated toolkit supporting tasks including automated phenotype extraction, data visualisation and automated model training. We validate PhenoAssistant through several representative case studies and a set of evaluation tasks. By lowering technical hurdles, PhenoAssistant underscores the promise of AI-driven methodologies to democratising AI adoption in plant biology. Application of large language models (LLMs) for automating complex and data-intensive crop phenotype analysis remains unexplored. Here, the authors integrate LLM with curated toolkit for phenotype extraction, visualization, and model training and show its ability to reduce technical barriers and enhance accessibility.
|
|
Scooped by
mhryu@live.com
April 3, 4:45 PM
|
Arbuscular mycorrhizal fungi (AMF) are key drivers of plant growth and nutrition, shaping the relationship between plant diversity and ecosystem productivity. In agroecosystems, AMF generally benefit crops but often have neutral or even negative effects on weeds, yet the mechanisms underlying these contrasting interactions remain poorly understood. In this Viewpoint, we propose a plant community-level framework to investigate interactions between multiple crop and weed species and diverse AMF taxa, focusing on chemically mediated communication via root exudates, particularly flavonoids (FLVs) and strigolactones (SLs). These compounds can act as ‘cry for help’ signals that recruit beneficial soil microorganisms to alleviate environmental stress. We found that their composition varies widely among plant families, with crops typically producing more diverse and functionally distinctive FLV profiles than weeds. Similar patterns, though less documented, appear for SLs. Different FLV subclasses elicit contrasting AMF responses, influencing spore germination, hyphal growth, and root colonization. Notably, FLVs with stronger positive effects on AMF are more common in crops, whereas those with neutral effects tend to dominate in weeds. Our results are consistent with the idea that such molecular cues may shape AMF recruitment and could potentially feed back into plant community dynamics, although this hypothesis should be explicitly tested.
|
|
Scooped by
mhryu@live.com
April 3, 4:26 PM
|
Nitrate accumulation is a prevalent challenge in aquaculture systems. Although aerobic denitrifiers offer a promising solution, few strains can tolerate varying salinities, and their performance in real aquaculture wastewater remains limited. In this study, an euryhaline aerobic denitrifying bacterium, Marinobacter sp. MAD1, was isolated from a recirculating aquaculture system. MAD1 demonstrated both ammonium assimilation and aerobic denitrification capacities, functioning effectively over a broad range of salinities (0–3.5%). Transcriptomic analysis suggests that strain MAD1 upregulates genes involved in K+ uptake and sulfur-containing amino acid metabolism, such as cysteine, to maintain cell growth and aerobic denitrification under hyposaline conditions. However, in seawater conditions, strain MAD1 survival relies on classical compatible solute accumulation. Bioaugmentation with culture MAD1 significantly improved nitrate removal compared to the control reactor under moderate (1.5%) and high salinity (3.5%), lowering effluent nitrate from 21.76 ± 3.14 to 3.58 ± 2.57 mg-N/L and 15.55 ± 2.69 to 4.51 ± 2.38 mg-N/L, respectively. Repeated amendment of strain MAD1 led to its predominance within the community, with relative abundances reaching 88.03% at 1.5% salinity and 81.09% at 3.5% salinity. Our findings highlight the potential of euryhaline aerobic denitrifiers for nitrogen management in aquaculture systems under variable salinity.
|
|
Scooped by
mhryu@live.com
April 3, 1:46 AM
|
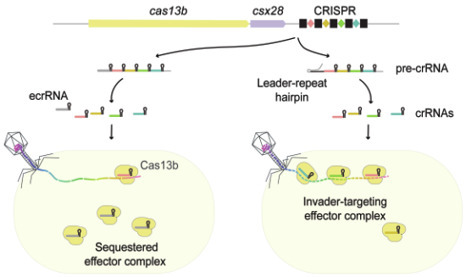
CRISPR RNAs (crRNAs) guide recognition and targeting of intracellular invaders as part of adaptive immunity by CRISPR-Cas systems. crRNAs are transcribed from CRISPR arrays of conserved repeats interlaced with invader-derived spacers. While crRNA production is essential for immunity, its optimization for defense remains poorly understood. Here, we show that, in diverse RNA-targeting type VI CRISPR-Cas systems, the leader RNA encoded upstream of the CRISPR array prevents formation of an invader-independent extraneous crRNA (ecrRNA) by blocking processing of the first repeat. Using the VI-B2 system from Porphyromonas gingivalis as a model, we demonstrate that the leader RNA and first repeat form a conserved inhibitory hairpin that precludes binding and processing by the system’s Cas13b nuclease. Disrupting this hairpin enables ecrRNA production, which in turn can deplete invader-derived crRNAs and reduce Cas13b-mediated phage defense. Structure prediction indicates that these leader-repeat hairpins are widespread across diverse type VI subtypes, highlighting a conserved regulatory mechanism. Our findings reveal how a prevalent branch of CRISPR-Cas systems suppresses ecrRNA formation to promote RNA-guided immunity.
|
|
Scooped by
mhryu@live.com
April 3, 12:54 AM
|
While the ocean’s photosynthetic production of organic matter rivals that on land, a combination of heterotrophy and sinking prevents significant accumulation of particulate organic matter (POM) in open ocean surface waters. The origins and fates of POM in ocean surface waters are unclear, in part due to the dominance of nonliving, altered material. From the natural nitrogen isotopic composition of chlorophyll and its degradation products, we estimate the fraction of particles from eukaryotic vs. prokaryotic phytoplankton. In subtropical gyres and along the eastern North Pacific margin, the eukaryotic-to-prokaryotic ratio in particles matches that of living phytoplankton. However, in the North Atlantic outside its subtropical gyre, particles have a lower eukaryotic-to-prokaryotic ratio than do the living phytoplankton. This discrepancy at least partly arises from preferential sinking of eukaryotic biomass, consistent with the canonical but disputed paradigm that cyanobacteria disproportionately fulfill the energetic demands of the upper ocean microbial community while eukaryotes drive export production. The prokaryotic bias in surface ocean particles may also result from slow decomposition of specific components of prokaryotic biomass, a possible bottleneck in the ocean’s microbial loop. The different fates of organic matter produced by eukaryotic and prokaryotic phytoplankton affect the productivity of the surface ocean, carbon export to the interior, and the signals recorded in deep-sea sediments.
|
|
Scooped by
mhryu@live.com
April 3, 12:34 AM
|
The stinkbug Plautia stali harbors essential gut symbiotic bacteria of the genus Pantoea, whose natural strains differ in cultivability and host benefits. Using this system, we evaluated how laboratory-evolved and genetically-engineered symbiotic E. coli strains compete against native Pantoea symbionts and how they influence host fitness. In single infection assays, the native uncultivable symbiont Sym A conferred the highest host performance, whereas the evolved (CmL05G13) and artificial (ΔcyaA) symbiotic E. coli strains supported host survival at levels comparable to cultivable Pantoea symbionts (Sym C-F). In competitive co-infection assays, the symbiotic E. coli strains generally showed unexpectedly strong colonization ability. CmL05G13 outcompeted all the cultivable symbionts Sym C-F and even displaced the native uncultivable symbiont Sym A, whereas ΔcyaA and the nonsymbiotic control E. coli ΔintS were dominated by Sym A at the adult stage. Despite their superior infection competitiveness, the symbiotic E. coli strains provided limited reproductive benefits, behaving as "cheater-like" associates. They were able to invade and dominate the symbiotic organ but failed to match the fitness contributions of native symbionts. These results demonstrate that the experimentally evolved E. coli can rapidly acquire strong colonization ability surpassing that of the natural symbionts that have coevolved with P. stali in nature. At the same time, the mismatch between infection success and host fitness benefits highlights potential evolutionary conflicts and provides an experimental model for studying the dynamics of cheating, mutualism, and symbiont replacement in vertically transmitted symbioses.
|
|
Scooped by
mhryu@live.com
April 3, 12:26 AM
|
Chemotaxis receptor complexes sense chemical gradient in the cellular environment to direct swimming towards favorable environments. The core signaling units of these complexes are made up of two trimers-of-dimers of chemoreceptors, two CheW and a CheA dimer, which further assemble into large hexagonal signaling arrays. Structural and biochemical studies have provided important information on the architecture and interfaces of these complexes. However, the signaling pathway of these complexes that controls the kinase is not fully understood. In this review we highlight the highest resolution models of this system and examine the current consensus on the protein-protein interfaces based on models and interface experiments. We also highlight differences observed between signaling states for the individual proteins and the protein interfaces that are proposed to be part of the signaling mechanism. Overall, we conclude that there is strong structural consensus for the protein interfaces but, despite some intriguing results, more information is needed to understand how the interfaces change between signaling states and the role they play in signaling. An animated Interactive 3D Complement (I3DC) is available in Proteopedia https://proteopedia.org/w/Journal:FEMS_Microbiology_Reviews:1.
|
|
Scooped by
mhryu@live.com
April 3, 12:18 AM
|
Understanding microbial communities requires moving beyond 2D representations toward a holistic view that couples 3D spatial organization with ecological function, integrating microbial inventories, genes, expression profiles, and interactions at scales and dimensions in which microbial life unfolds. In this opinion article, we synthesize recent findings and emerging approaches that enable the investigation of microbial interactions within their native 3D context. We propose conceptual frameworks for integrating spatial–functional information into comprehensive ecological maps, providing new avenues to interpret microbial interactions and to test ecological theory in situ. Together, these insights outline a new ecological paradigm for microbiome research and highlight how spatially resolved understanding can be harnessed to interpret and ultimately guide the modulation of microbial interactions and ecosystem function in natural settings.
|
|
Scooped by
mhryu@live.com
April 2, 11:52 PM
|
Bioactive natural products are promising for diverse applications, prompting their efficient synthesis in microbial cell factories, enabled by advances in synthetic biology. However, the metabolic burden caused by the intracellular accumulation of natural products impairs cell growth and limits final production. Therefore, relieving the metabolic burden of microbial cell factories has emerged as an attractive strategy to enhance the production of natural products. Here, we focus on the recent advances in the yeast species Saccharomyces cerevisiae, Yarrowia lipolytica, and Rhodosporidium toruloides for enhancing the synthesis of natural products (with a focus on terpenes) by relieving the metabolic burden from intracellular accumulation. Three strategies to lower this metabolic burden are reviewed: 1. overexpressing and engineering transporters, 2. changing membrane structure, and 3. In situ extraction. Future directions in natural product export are summarized and may help reify efficient, sustainable, and economically viable natural product synthesis.
|
































m-2st, The cargo protein of interest is fused to a streptavidin-binding peptide (SBP) tag and co-expressed with a “hook” protein that contains a streptavidin (Strep) domain anchored to a specific organelle. In the absence of biotin, the SBP-cargo is retained via the SBP-Strep interaction. Addition of biotin competitively disrupts this interaction, leading to synchronous release and trafficking of the cargo and allowing monitoring of trafficking and secretion in real time. an SBP-tagged cargo reversibly binds a streptavidin-tagged hook localized to a defined organelle. Addition of biotin disrupts the streptavidin-SBP interaction, releasing the cargo. HiBiT-SBP-TNFα, -Tau, or -IL1β were transduced with lentivirus constructs encoding hooks targeted to the ER lumen (Strep-KDEL) or to the cytosol-facing side of the ER membrane (Ii-Strep).