 Your new post is loading...

|
Scooped by
mhryu@live.com
Today, 2:02 AM
|
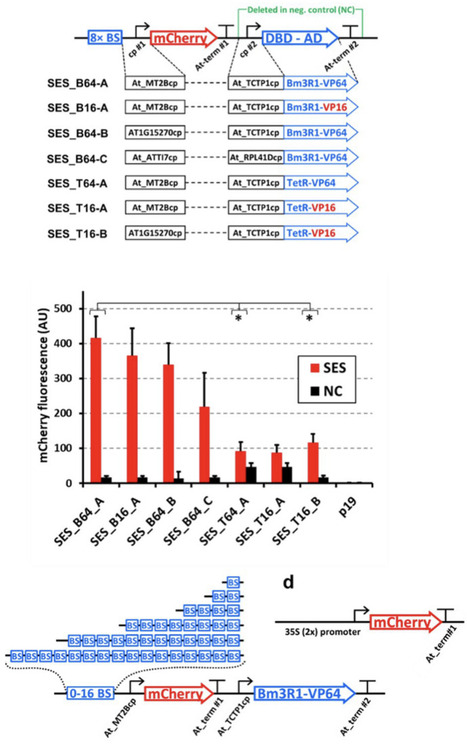
Plants represent commercially relevant production systems for recombinant proteins and chemical compounds. Effective genetic engineering depends on precise control of heterologous gene expression, which remains challenging due to complex transcriptional and post-transcriptional regulation, endogenous gene silencing mechanisms, and notably because of limited number of tools for allowing robust, fine-tuned control of expression levels across different systems/organisms. Some of the most common issues associated with plant expression systems are addressed with our plant-optimized version of a previously developed fungal universal synthetic expression system (SES). Plant SES demonstrates several favorable characteristics for robust heterologous gene expression, including highly constitutive function with apparently reduced sensitivity to endogenous silencing in transient assays, without requiring p19 co‑expression under the tested conditions. These together provide simple, predictable tuning of gene expression levels, and the potential for very high expression levels of the target genes. In all these features, SES shows higher and more stable transcript levels than Cauliflower Mosaic Virus (CaMV) 35 S promoter-based constructs in our experimental setups. The functionality of plant SES was tested by expressing mCherry and three commercially relevant proteins: fungal glucose oxidase (GOX), protein A and human vascular endothelial growth factor 165 (VEGF16) from diverse organisms, supporting high-level accumulation of recombinant proteins. In addition, plant SES retains full functionality in both plant and fungal hosts, which makes this expression system a useful tool for a multitude of genetic engineering applications in other eukaryotic organisms.
|
|
Scooped by
mhryu@live.com
Today, 1:53 AM
|
Plasmids benefit bacterial communities by storing auxiliary genes that address environmental challenges such as antibiotics. Subsequent plasmid loss can also be advantageous if plasmid benefits are temporary but costs are permanent. However, unless positive selection is sustained, plasmid loss can proceed to extinction, with access to plasmid-derived benefits permanently lost. In principle, horizontal transmission can maintain a plasmid in a population, but if the plasmid cost is too high, the host can become uncompetitive. We examine how survival of costly but occasionally beneficial plasmids is possible in a bacterial population. Using population models, we demonstrate that plasmid-dependent phages can, counterintuitively, solve this plasmid survival problem for their bacterial hosts. Phage predation pins the plasmid at low but nonzero abundance, such that the plasmid cost is effectively neutralized at the population level, dramatically lengthening the persistence time of the plasmid. When conditions change and the costly plasmid becomes beneficial, it spreads across the host population and switches to a vertical-transmission lifestyle until benefits again subside.
|
|
Scooped by
mhryu@live.com
Today, 1:25 AM
|
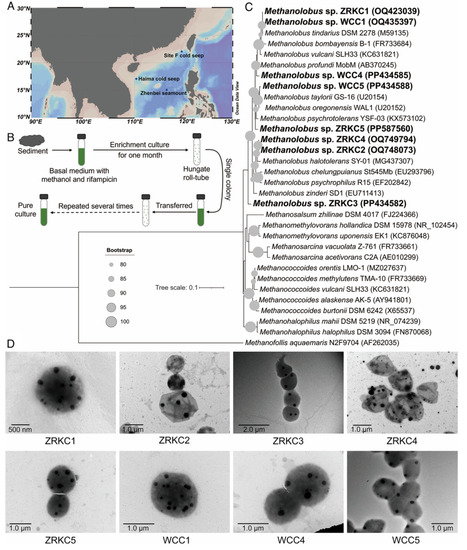
Microorganisms employ inorganic polyphosphate (polyP) as an ancient strategy for energy and phosphate storage, yet its physiological roles in methanogenic archaea remain largely unexplored. Here, we report that polyP metabolism is coupled to growth and methanogenesis in Methanolobus sp. ZRKC1, a representative of eight methanogenic archaea isolated from deep-sea cold seep sediments. Through combined genetic, biochemical, and physiological analyses, we find that PPK1 mediates polyP synthesis in a Mg2+-dependent manner, whereas PPK2 functions primarily as a polyP hydrolase. Deletion of ppk1 in strain ZRKC1 abolishes polyP accumulation and impairs both growth and methane production, pointing to a role for polyP as a metabolic hub linking these processes. Transcriptomic profiling reveals that under organic phosphorus conditions, strain ZRKC1 upregulates phosphoesterases to liberate bioavailable phosphate, which is subsequently channeled into polyP via PPK1. Furthermore, in situ transcriptomic data suggest that the genetic capacity for this metabolic strategy may be present and transcriptionally active in the native environment, with concurrent upregulation of genes involved in phosphate acquisition, polyP metabolism, and methanogenesis. Our findings suggest the importance of polyP in linking phosphate homeostasis to growth and methanogenesis in deep-sea methanogenic archaea.
|
|
Scooped by
mhryu@live.com
Today, 12:45 AM
|
Studies of whole viral populations—the “virome”—are yielding exciting new insights into biological systems, but methods are still being optimized. Here, we describe generation and use of a synthetic viral community and its use to evaluate technical challenges arising in virome analysis. We spiked the mock community into different human sample types, then passed the samples through different virus enrichment protocols and analyzed by Illumina sequencing. Compared with direct metagenomic sequencing, VLP enrichment protocols greatly increased viral read yields from stool and saliva. Four methods for DNA amplification were compared, with three showing over-amplification of small circular ssDNA viruses, most notably GenomiPhi. Studies of viral particle stability in the presence of nuclease showed that most viral genomes were stable when protected in viral particles, but phage MS2 RNA was unexpectedly labile under some of the conditions tested. Comparison of Illumina 1,000-cycle sequencing versus 300-cycle sequencing showed that longer reads supported generation of longer viral genome assemblies. We tested bacteriophage T4 DNA modified with glucosyl-hydroxymethylcytosine (ghmC) and hydroxymethylcytosine (hmC) and found that both were readily detected, though the recovery of ghmC-modified DNA was reduced compared with T4 genomes with unmodified cytosine. These studies together with published data help provide guidance for virome researchers optimizing analytical protocols.
|
|
Scooped by
mhryu@live.com
Today, 12:34 AM
|
Connecting mathematical models with empirically measured microbial growth has remained challenging, as numerous competing models based on different theoretical approaches can fit observations. Therefore, we develop a method to automatically propose growth models from microbial data alone. We validate this approach using an available dataset of E. coli grown on known resources, and study 14 species across various concentrations of a rich medium. The inherently interpretable approach of symbolic regression infers explicit dynamical models directly from growth data. Using symbolic regression natively, does not favor biologically interpretable models, but we find cumulative population gain to be a more informative machine learning feature than population size. Random Forest machine learning allows us to relate this finding to the approximation of a constant-rate per capita resource consumption. This suggests that the area under the growth curve (AUC) measured in routine experiments provides information on the effective resource dynamics governing microbial growth. Finally, we use theoretical insights to inform the symbolic regression algorithm and favor biologically interpretable models. Overall, we found that balancing between data fit, parsimony and biological relevance favored both the simplest, linear approximation and models based on Monod dynamics, with either one or two underlying resources. Therefore, our approach to read growth laws off of microbial batch cultures provides insights on data-driven modelling.
|
|
Scooped by
mhryu@live.com
Today, 12:04 AM
|
Environmental samples contain complex microbial communities hiding a treasure trove of active compounds. However, screening for active natural products from environmental samples is challenging due to inefficient cultivation techniques and a lack of proper screening platforms. For empowering antibiotic screening assays from complex microbial communities, we have developed and optimized a droplet-based platform with multiplexing capability. A cultivation strategy for bacteria in picoliter droplets was combined with phenotypic screening using multiple whole-cell reporter species. A mixture of two different fluorescently labelled reporter strains, one Gram-positive and one Gram-negative, was picoinjected to each of millions of picoliter cultures, which were screened for inhibiting activity based on the independent survival signals of each reporter species. Proof-of-concept experiments demonstrate efficient detection, selection, and recovery of a model Streptomyces strain from a synthetic mixture according to the specific inhibition of the reporter strains by the produced antibiotic. Subsequently, the established platform was successfully applied to screen environmental microbial communities from soil samples. This approach showcases multiplexing capabilities for screening assays in microfluidic droplets in order to simultaneously screen for new bioactive compounds with various inhibition profiles.
|
|
Scooped by
mhryu@live.com
June 2, 11:27 PM
|
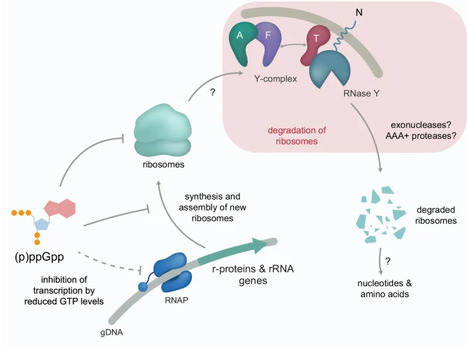
Limiting ribosome synthesis and activity is crucial for adaptation to stresses, such as heat or nutrient starvation. In Bacillus subtilis, this can be achieved through the coordinated action of the alarmones (p)ppGpp and the transcription factor Spx. Here, we performed a genetic screen to identify novel factors that contribute to the heat shock response in B. subtilis. We identified the Y-complex, which confers specificity to the endonuclease RNase Y, as a critical player under stress conditions, such as heat or transition into the stationary phase. This protein complex is required for the targeting and processing of diverse RNAs, notably the maturation of mRNAs encoding proteins involved in translation and metabolism. We further demonstrate that the Y-complex and RNase Y initiate the degradation of rRNAs of mature ribosomes, lowering their abundance. We propose that the Y-complex is a regulatory hub that modulates gene expression, adjusts protein synthesis and resource allocation. Transcriptional regulation is a central mechanism underlying bacterial adaptation to stress. Here, the authors used a genetic fitness screen to show that the Y-complex in Bacillus subtilis contributes to the heat shock response through its interaction with the ribosome.
|
|
Scooped by
mhryu@live.com
June 2, 6:08 PM
|
Protein-based fluorescence imaging is a powerful modality for visualizing diverse biological processes. Biological imaging in the near-infrared (NIR, 800–1000 nm) and shortwave infrared (SWIR, 1000–2000 nm) ranges confers a number of photophysical advantages, but remains a challenge in practice due to the dearth of suitable protein probes in these optical windows. To address this limitation, we sought to develop a general approach integrating computational protein design with organic synthesis for creating long-wavelength fluorescence-activating proteins from scratch. We used this approach to de novo design proteins that specifically bind to synthetic merocyanine dyes, forming Schiff base covalent linkages, which when protonated activate fluorescence with large redshifts in both excitation and emission wavelengths. We describe a designed far-red fluorescence-activating protein, MC7BP34, with a brightness greater than that of existing fluorescent proteins in a similar wavelength range, and an NIR design MC9BP81 with excitation at 892 nm and emission extending into the SWIR range with higher contrast and imaging sensitivity in vivo than the previously developed iRFP720 (excitation 672 nm) owing to the reduced tissue autofluorescence at longer wavelengths. Our results are a substantial step toward genetically encodable probes in the SWIR region, and our approach lays the groundwork for the development of NIR biosensors for specific biological applications.
|
|
Scooped by
mhryu@live.com
June 2, 4:13 PM
|
Harmful cyanobacterial blooms are increasing worldwide, threatening freshwater ecosystems, animal health, and human well-being. To guide research needed for effective prediction, prevention, and management, we identify four priorities: understanding eco-evolutionary and phylogeographic drivers that promote toxigenic cyanobacterial genotypes; resolving molecular and environmental controls on cellular cyanotoxin biosynthesis; integrating microbiome science with multiomics and epidemiology to assess associated health risks beyond cyanotoxins; and quantifying ecosystem-scale bottom-up and top-down controls in understudied settings, particularly tropical and benthic habitats. New research tools for addressing these priorities enable elucidating the mechanistic basis for anticipating cyanobacterial blooms and provide the understanding needed for their control in a rapidly changing world.
|
|
Scooped by
mhryu@live.com
June 2, 4:08 PM
|
N-linked glycosylation is critical for protein function and stability, yet identifying glycosylated sites remains challenging because glycosylation depends on sequence motifs and structural context. Many available computational approaches focus on motif-centred sequence windows and provide limited support for whole-protein inspection of candidate sites. SGGly is a freely accessible web server for structure-guided analysis of candidate N-linked glycosylation sites across full-length proteins. The server uses ProtBERT transformer-based embeddings with sequon and structure-derived residue descriptors to generate residue-level candidate-site predictions and returns downloadable residue-level predictions together with interactive 3D visualisation. Using a dual evaluation framework, SGGly achieved a Matthews correlation coefficient of 0.888 and receiver operating characteristic area under the curve of 0.987 under a strict, publication-supported regime. On the independent N-GlyDE benchmark, SGGly demonstrated strong generalisability, achieving the strongest specificity (0.941), sensitivity (0.993), and accuracy (0.946) among compared methods. SGGly provides a practical web resource for whole-protein glycosylation candidate mapping, structural inspection, and prioritisation of sites for follow-up analysis, guiding experimental design and interpreting glycoproteomic observations.
|
|
Scooped by
mhryu@live.com
June 2, 3:55 PM
|
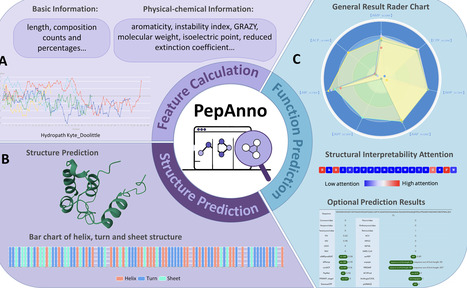
Peptides are gaining prominence as therapeutic candidates due to their diverse physiological functions and structural simplicity. Although multiple computational tools exist for bioactive peptide prediction, many suffer from limitations such as non-intuitive interfaces, sequence-only representations, insufficient structural awareness, restricted interpretability, or fragmented analysis workflows, leading to reduced research efficiency and higher costs. To address these challenges, we present PepAnno (https://bis.zju.edu.cn/pepanno/), a comprehensive and user-friendly web server for multi-functional peptide annotation. PepAnno is powered by a novel structure-aware, multi-view geometric deep learning framework that integrates pre-trained sequence embeddings with predicted 3D structural graphs through a dual-stream architecture combining a Transformer and a GATv2 network. A cross-modal attention mechanism is employed to effectively fuse semantic and geometric representations, enabling accurate multi-task prediction across 7 key bioactivities, including antimicrobial and anticancer properties. Comprehensive evaluation on seven curated bioactivity datasets demonstrates that PepAnno achieves robust and competitive predictive performance across tasks, consistently outperforming or matching existing methods in terms of discrimination and stability. Beyond functional prediction, PepAnno provides automated calculation of physicochemical properties, structure visualization, and access to an integrated repository of peptide-related databases and tools. By enabling one-click peptide annotation, PepAnno offers an efficient and interpretable solution for large-scale peptide analysis and facilitates downstream experimental design and peptide-based drug discovery.
|
|
Scooped by
mhryu@live.com
June 2, 3:34 PM
|
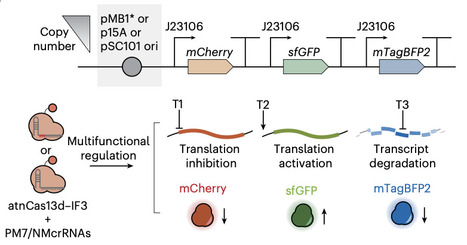
Cas13-based RNA effectors may enable dynamic, multiplexed and reversible gene regulation in bacteria. Yet, their widespread adoption is hindered by inherent cytotoxicity and collateral cleavage. Here we present a rational protein engineering strategy to generate attenuated Cas13d variants with tunable RNase activity through targeted truncation of flexible regions. This permits effective transcript knockdown while greatly reducing toxicity as reflected by a 2.2-fold higher growth optical density. By introducing proximal mismatches at the 5′ end of CRISPR RNA spacers, our system allows functional switching between translation inhibition, polycistronic mRNA degradation and IF3-fusion-based translation-level CRISPR activation. We demonstrate programmable, orthogonal and multiplexed regulation of individual genes within polycistronic mRNAs and synthetic circuits. Application to lycopene biosynthesis optimization shows robust pathway rewiring and improved yields alongside fine-tuned modulation of essential and competing pathways in E. coli. Our work provides a versatile RNA-regulatory toolkit for next-generation microbial synthetic biology and RNA-based biotechnology. A Cas13-based gene switch allows multiplexed RNA regulation in bacteria.
|
|
Scooped by
mhryu@live.com
June 2, 3:11 PM
|
Symbiotic bacteria in the gut have an important physiological impact on their hosts, but the mechanisms that underlie the exchange of molecular signals remain poorly understood. Membrane vesicles have been suggested to mediate the direct exchange of cytoplasmic content like nucleic acids (NAs), but their study is complicated by conflicting and imprecise reports of their type and composition. Here, we show that honeybee gut symbionts produce non-lytic membrane vesicles (MVs) enriched in NA, potentially explaining the RNAi activity of engineered Snodgrassella alvi in honeybees. Using cryogenic electron microscopy (cryo-EM), we developed a method to distinguish lytic from non-lytic MV production in Gram-negative bacteria and to differentiate outer membrane vesicles (OMVs) from outer-inner membrane vesicles (OIMVs) based on membrane ultrastructure. Among the strains studied, S. alvi, Gilliamella apicola, and Gilliamella apis exhibit clear OMV and OIMV budding, while Escherichia coli and Salmonella enterica show membrane debris and self-assembled vesicles, indicating lytic release. MVs from the symbionts carry significantly more NAs than non-symbionts. Assays on DNA and RNA contents confirm the cytoplasmic origin of MV cargo in S. alvi, suggesting a role in mediating NA delivery to the insect host. These findings enhance our understanding of symbiotic vesiculation and highlight the potential for engineering symbionts to boost honeybee immunity and deliver NA-based therapeutics via vesicular transport. Here, using cryo-EM, the authors visualize non-lytic outer and inner membrane vesicles budding from Gram-negative symbionts, and provide evidence that these nucleic acid-rich double membrane vesicles may transport and deliver cytoplasmic cargo to the host.
|
|
|
Scooped by
mhryu@live.com
Today, 1:58 AM
|
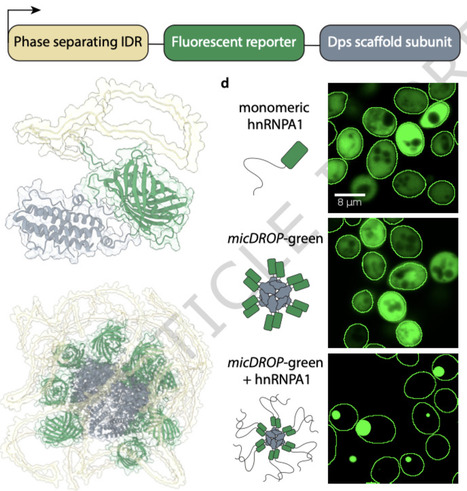
Intrinsically disordered regions (IDRs) drive intracellular phase separation and biomolecular condensate formation through interactions encoded in their sequence. Although condensates form spatially distinct assemblies in cells, the high conformational flexibility of IDRs and absence of well-defined 3D structures raise the question of how they could encode condensate specificity. To systematically characterize IDR–IDR interactions and their ability to mediate self-specific partitioning, we develop micDROP, a synthetic system of multivalent IDRs forming constitutive droplets. We examine ten natural IDRs that phase-separate in micDROP and find that their saturation concentrations in vivo correlate with total sequence stickiness. Co-expression of IDR pairs fused to distinct micDROP scaffolds reveals widespread promiscuity, whereas TDP43 and UBQ2 consistently form self-specific condensates. A short, conserved α-helical segment in the TDP43 IDR governs this self-recognition. Our results indicate that IDRs tune phase separation propensity through sequence composition, while discrete condensate identity likely requires additional structural determinants. Here the authors develop micDROP to systematically test IDR-IDR interactions driving condensate formation. Most IDR pairs co-mix promiscuously; only TDP43 and UBQ2 form distinct droplets, with a short α-helix in TDP43 driving self-recognition.
|
|
Scooped by
mhryu@live.com
Today, 1:51 AM
|
Genomovar-level and intragenomic diversity cannot be resolved by conventional amplicon sequencing due to the limitation of fragment lengths and read accuracy, while the application of metagenomic profiling to a large number of samples can be resource intensive. Here, we report UltraRes-rrn, an integrated wet-lab and computational workflow for high-accuracy rrn (i.e., 16S-ITS-23S rRNA) operon profiling using Nanopore sequencing. By integrating ultra-long DNA recovery, long-read amplicon sequencing, and unique molecular identifiers (UMI)-based consensus correction, UltraRes-rrn obtains full-length 16S-ITS-23S rRNA operon consensus sequences with mean accuracies exceeding 99.98%. To achieve higher resolution, we propose a hierarchical rRNA operon profiling strategy in which concatenated 16S+23S rRNA genes (16S23S) serve as a primary and the internal transcribed spacer (ITS) provides a secondary marker. The 16S23S marker achieved discrimination at the genomovar level compared to either 16S or 23S rRNA genes alone, which mitigates the ITS-driven over-splitting observed with the full-length rrn operon and allows for larger proportion of data being classified at higher confidence thresholds. Further, ITS variation was strongly structured by tRNA occurrence patterns, suggesting that ITS can capture taxon microdiversity missed by either 16S or 23S rRNA gene sequences alone. The UltraRes-rrn workflow was applied to full-scale nitrogen removal reactors, revealing intraspecies diversity variation driven by different carbon regimes, which would not have been possible with a shorter gene sequence. In summary, UltraRes-rrn enables cost-effective community profiling at genomovar-level resolution in complex ecosystems.
|
|
Scooped by
mhryu@live.com
Today, 12:59 AM
|
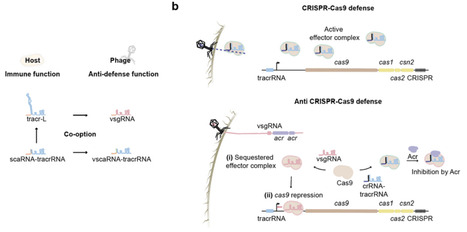
Central to CRISPR technologies is the single-guide RNA (sgRNA), an engineered fusion of a processed CRISPR RNA and tracrRNA that directs Cas9 and many Cas12 nucleases to bind and cleave target DNA. Here, we report the discovery of similarly compact viral sgRNAs (vsgRNAs) encoded by bacteriophages that counteract bacterial CRISPR-Cas9 immunity. vsgRNAs inhibit Cas9 function via two complementary routes: by sequestering the Cas9 apoenzyme, and by re-directing the Cas9 nuclease to transcriptionally silence its own promoter. vsgRNAs also can cooperate with co-encoded anti-CRISPR proteins (Acrs), including AcrIIA25.1 that blocks DNA binding by Cas9 complexed with a standard sgRNA but not with the vsgRNA. We predict that phages evolved vsgRNAs by co-opting and repurposing host-encoded long-form tracrRNAs (tracr-L) responsible for Cas9 auto-repression and a countermeasure to Acrs. Our search also uncovered Cas9-regulating small CRISPR-associated RNAs (scaRNAs), which we predict were inserted upstream of tracrRNAs to form tracr-L but were also co-opted by phages as viral scaRNAs to suppress Cas9 immunity. Finally, we found that vsgRNAs can enable genome editing in mammalian cells, offering a natural guide RNA template for CRISPR technologies. Overall, these findings reveal that bacteriophages devised their own compact sgRNAs tailored to subvert Cas9 immunity, long preceding their rational design to program RNA-guided nucleases.
|
|
Scooped by
mhryu@live.com
Today, 12:37 AM
|
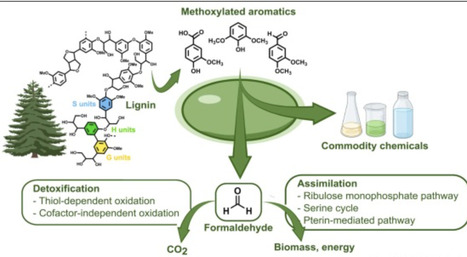
Formaldehyde is a toxic metabolite generated in the bacterial catabolism of lignin-derived aromatic compounds (LDACs) through the oxidative O-demethylation of methoxylated aromatics. Microbial cell factories designed to convert LDACs to commodity chemicals should ideally utilize formaldehyde to improve carbon efficiency and strain fitness. Methylotrophs and natural degraders of LDACs have evolved various strategies for utilizing formaldehyde. These range from cofactor-dependent detoxification systems that extract reducing equivalents to cyclical pathways that synthesize building blocks. In addition, recent engineering efforts, driven in part by interest in C1 feedstocks, have yielded useful synthetic formaldehyde metabolic pathways. In this review, we outline several of these routes, highlighting gaps and metabolic engineering opportunities, with a particular focus on improving LDAC biocatalysts.
|
|
Scooped by
mhryu@live.com
Today, 12:23 AM
|
The ever-expanding catalogue of uncharacterized proteins - the so called functional dark matter - poses a major challenge for biotechnological and biomedical exploitation. Functional assessment of most proteins is hindered by the technical limitations of annotation transfer and by the propagation of erroneous annotations in databases. The common denominator here is the reliance on sequence similarities. However, these become inaccurate below certain thresholds and can diverge even at sequence identities around 70%. To approach this challenge, we implemented a strategy using embeddings generated by protein language models for targeted function discovery (PE-TFD). Datasets of proteins representing target as well as non-target functions were used to train supervised learning models. The resulting ensemble models yielded interpretable prediction scores, enabling the exploration of databases without relying on multiple sequence alignments or structural information. We here tested PE-TFD for the discovery of novel hydrogenases as proof-of-concept, resulting in the novel discovery of 773 [NiFe] and 1,929 [FeFe] hydrogenases that were not detected by established sequence- or profile-based approaches. Structural analyses supported their non-random nature and further revealed a significant number of enzymes lacking prior functional annotation. Our framework therefore enables interpretable function discovery in large-scale datasets and the exploitation of functional dark matter.
|
|
Scooped by
mhryu@live.com
June 2, 11:32 PM
|
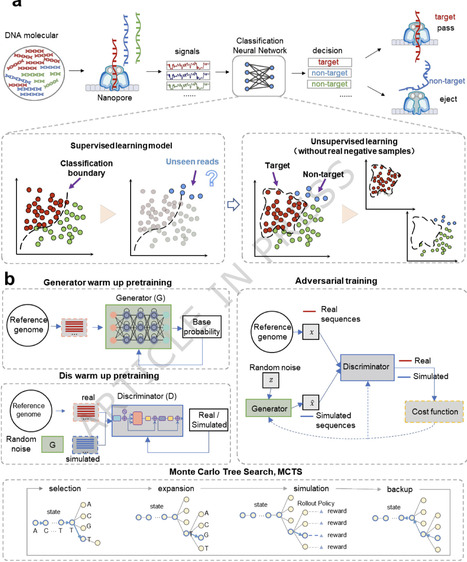
Nanopore adaptive sequencing enables real-time target enrichment, yet current deep-learning methods require costly, sample-specific experimental training data. To address this, we developed GANBase, a genome-guided generative adversarial learning framework, which is trained exclusively on reference sequences and incorporates a Monte Carlo Tree Search-based Rollout strategy for model training. GANBase demonstrates robust performance in target enrichment and host depletion across diverse scenarios. In live adaptive sequencing experiments, it remains effective despite significant pore loss or flow cell version updates, providing a data-independent solution that significantly expands the utility of real-time targeted sequencing. Adaptive nanopore sequencing enables targeted enrichment, but current methods rely on training data. Here, the authors develop GANBase, a genome‑guided deep‑learning framework that achieves robust, data‑independent target enrichment and host depletion across diverse sequencing conditions.
|
|
Scooped by
mhryu@live.com
June 2, 11:02 PM
|
Non-conventional yeasts offer unique advantages as tailored cell factories. Pichia pastoris is a master of protein expression and secretion. However, engineering P. pastoris for the overproduction of secondary metabolites is challenging due to its tightly regulated metabolism. How can we overcome this metabolic rigidity for efficient biosynthesis of energy-demanding molecules such as sclareol, a valuable diterpenoid? In this research article, we developed a host-specific framework integrating systematic pathway-level rewiring of the sclareol biosynthetic pathway and central metabolism to alleviate precursor acetyl-coenzyme A and cofactor NADPH limitations. Regulatory-level remodeling identified metabolic factors that enhance cellular robustness and protein homeostasis, reprogrammed global transcriptional regulators for carbon and nitrogen flux balance, and tailored process-level fermentation to eliminate byproducts. These efforts enabled a sclareol titer of 27.8 g l−1 in minimal medium, representing the highest diterpenoid production reported. This work redefines P. pastoris as a powerful chassis for natural product biosynthesis, providing a generalizable roadmap for engineering non-conventional hosts.
|
|
Scooped by
mhryu@live.com
June 2, 6:01 PM
|
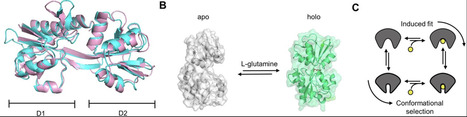
The glutamine-binding protein GlnBP is part of an ATP-binding cassette transporter system in E. coli and uses two well-characterized conformational states, an open ligand-free and a closed-liganded state, to facilitate active amino-acid uptake. Existing literature on its ligand-binding mechanism lacked sufficient evidence to univocally assign the kinetic type of binding mechanism for GlnBP: ligand binding prior to conformational change, that is an induced fit, or the conformational selection, in which the ligand binds the matching conformation from a pre-existing ensemble. Since such mechanistic questions are relevant for our fundamental understanding of how this and other biomacromolecules regulate cellular processes, we here revisit the question for GlnBP. We present a biochemical and biophysical analysis using a combination of calorimetry, single-molecule and surface-plasmon resonance spectroscopy, and molecular dynamics simulations. We found that both apo- and holo-GlnBP show no detectable exchange between open and (semi-)closed conformations on timescales between 100 ns and 10 ms and that ligand binding and conformational changes in GlnBP are correlated. A global analysis of our experimental results suggests that the conformational selection model is only compatible with GlnBP for the extreme scenario of very fast conformational exchange between the open and closed states on timescales <100 ns. In contrast, all data remains compatible with an induced-fit mechanism, where the ligand binds GlnBP prior to conformational rearrangements. Importantly, our work demonstrates that it is an intricate task to identify the type of kinetic binding mechanism and that this requires not only a sufficient set of data, but also an integrative experimental and theoretical framework to address the question. Based on this concept, we propose that various protein systems, for which so far only insufficient kinetic data are available, should be revisited.
|
|
Scooped by
mhryu@live.com
June 2, 4:10 PM
|
New approaches for molecular analysis continuously open new vistas in molecular medicine. Our lab has built a series of molecular detection techniques based on enzymatic ligation of synthetic oligonucleotides as versatile tools to gain new molecular insights. These fundamental techniques continue to yield new means for specific, high-throughput analyses of nucleic acids and proteins in contexts of relevance for molecular medicine. The combined potential for vast multiplexing and low sample consumption renders the assays described herein attractive as a basis for AI-assisted model building in medicine. Accordingly, this overview is aimed to present a ligase-based molecular toolbox to choose from in addressing present and upcoming analytical needs.
|
|
Scooped by
mhryu@live.com
June 2, 4:03 PM
|
Exploring the dynamical and structural properties of molecular complexes involving DNA is a fundamentally important aspect of understanding many biological processes. Although tools exist for modeling linear DNA and simple complexes, significant challenges remain in generating intricate biomolecular assemblies and incorporating biologically relevant modifications. These limitations restrict the ability to create accurate starting configurations for advanced molecular simulation studies. Here, we introduce MDNA, a molecular modeling toolkit that bridges these gaps by enabling the construction and analysis of complex DNA structures. MDNA provides a versatile solution to generate DNA shapes using a spline-based mapping technique that enables the construction of DNA configurations with arbitrary shapes. Key features include support for (non-)canonical base modifications, such as Watson–Crick–Franklin to Hoogsteen transitions, DNA methylation, and the ability to refine structures using Monte Carlo minimization. The toolkit also provides geometric analysis tools based on rigid body formalism to evaluate DNA structures and trajectories. Together, these features enable users to model and analyze DNA configurations in high detail with a modular Python interface. By integrating structure generation and analysis into a single workflow, MDNA facilitates the study of DNA–protein interactions, supporting new insights into DNA dynamics and molecular simulations.
|
|
Scooped by
mhryu@live.com
June 2, 3:49 PM
|
Multidrug-resistant bacteria necessitate innovative antibacterial strategies. Bacteriophages (phages) offer a promising alternative; however, bacterial immune defenses limit their effectiveness. Small-molecule inhibitors of these defenses may facilitate mechanistic studies and serve as adjuvants to enhance phage therapy. Here, we identify inhibitors targeting the bacterial cyclic oligonucleotide-based anti-phage signaling system (CBASS) effector nuclease Cap5. Cap5 is hypothesized to degrade genomic DNA in virally infected cells, leading to cell death through abortive infection. Guided by the crystal structure of the Cap5 SAVED domain bound to its activating ligand, we performed structure-guided virtual screening to identify candidate inhibitors. Biochemical assays revealed ∼16% of the top docking hits inhibited Cap5. Cellular assays revealed one compound could enter E. coli cells and inhibit Cap5 activity. Our integrated approach—combining structure-based virtual screening with biochemical validation—provides a framework for discovering small-molecule inhibitors of bacterial immune defenses to advance adjunctive therapies and deepen our understanding of phage-bacteria interactions.
|
|
Scooped by
mhryu@live.com
June 2, 3:17 PM
|
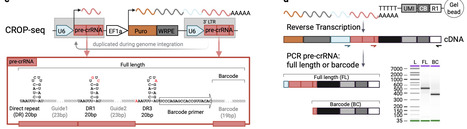
Single-cell perturbation (Perturb-seq) screens have primarily relied on Cas9 for inducing loss-of-function phenotypes, whereas Cas12a, despite its unique effectiveness for multiplex guide expression, remains underexplored. This may be due to Cas12a’s guide RNA array (pre-crRNA) self-processing activity and the subsequent challenges associated with pre-crRNA sequence recovery during single-cell RNA sequencing library preparation. To overcome the self-processing constraint, we optimized pre-crRNA expression vectors and established a degron-based, enhanced Cas12a system for gene knock-out. As demonstrated across cell types, target genes, and with a minimized guide RNA library, this platform allows for accurate detection of pre-crRNAs and gene editing-induced effects on the transcriptome in single cells. Additionally, we show that HyperLbCas12a outperforms other existing variants for multiplexed gene suppression. While the rapid reversibility of this repressor highlights specific kinetic constraints for degron-based single-cell recording, the system provides a potent, modular tool for contexts requiring tunable, transient silencing. Together, this suite of technologies greatly expands the possibilities for future Perturb-seq efforts and broader application of Cas12a for genetic disruption at scale. Cas12a self-processing of guide RNA transcripts limits its use for Perturb-seq by preventing guide capture. Here, authors develop a chemically degradable Cas12a platform enabling accurate guide assignment in gene knockout experiments and potent multiplexed, transient gene suppression.
|
























r-3st, Vasculature-derived amino acid leakage occurs at specific root sites, including pre–Casparian strip (CS) formation regions, lateral root emergence (LRE) sites, and CS-defective areas caused by endodermal cell damages or genetic defects. This leakage generates a strong chemotactic signal for bacteria in the rhizosphere, promoting their accumulation and proliferation at specific regions of the root.
Our observation that bacterial colonization patterns coincide with absent or impaired endodermal barriers led us to hypothesize that transient endodermal leakages of organic compounds might drive bacterial attraction and colonization of roots