 Your new post is loading...

|
Scooped by
mhryu@live.com
Today, 2:55 PM
|
During maize (Zea mays L.) domestication, seed protein content sharply declined. In plants, glutamine and asparagine levels are closely correlated with protein content. Asparagine is synthesized from glutamine, a process catalysed by asparagine synthase. Teosinte harbors a superior haplotype of asparagine synthase 4 (ASN4). Here, we report that teosinte also possesses a superior haplotype gene promoting glutamine synthesis. We identify and clone teosinte high protein 3 (THP3), which encodes glutamate-oxaloacetate transaminase 1 (GOT1), a key enzyme involved in nitrogen assimilation and carbon–nitrogen balance. The superior THP3-T allele, subjected to negative selection during domestication, has natural variations that boost both its expression and enzymatic activity. Overexpressing THP3-T, but not the modern THP3-B allele, significantly increases seed protein, representing altered carbon–nitrogen composition. Pyramiding THP3-T with THP9-T (the latter encoding asparagine synthase 4 (ASN4)) synergistically elevates both seed and whole-plant protein in elite hybrids while maintaining yield. Our findings demonstrate a powerful strategy for crop improvement by reintroducing beneficial rare alleles disfavoured during domestication from wild relatives. Teosinte alleles enhance nitrogen assimilation and seed protein without lowering crop yield when expressed in modern maize, providing a powerful strategy for crop improvement to meet future population demands.
|
|
Scooped by
mhryu@live.com
Today, 2:29 PM
|
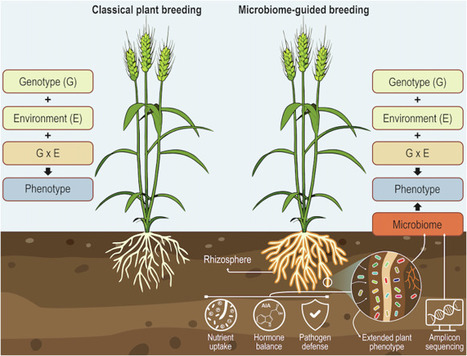
Plant breeding has advanced through genomics and predictive models, yet the plant-associated microbiome remains largely excluded from plant breeding trials. Microbial communities strongly influence nutrient acquisition, stress tolerance, and disease resistance, shaping key agronomic traits. We argue that neglecting microbiome variation biases heritability and G×E estimates, constraining genetic gains. Integrating microbiome information into breeding trials offers a feasible, scalable path toward more predictive, resilient, and sustainable crop improvement.
|
|
Scooped by
mhryu@live.com
Today, 2:18 PM
|
Antimicrobial resistance (AMR), particularly Gram-negative bacteria, poses significant challenges due to their robust outer membranes limiting antibiotic efficacy. Antimicrobial peptides (AMPs) show promising potential to replace traditional antibiotics. This study proposes a multi-condition constrained directed generation framework guided by the AMP antimicrobial mechanisms for designing membrane-targeting antimicrobial peptides (MTAMPs) against Gram-negative bacteria. By integrating sequence information, physicochemical properties, and spatial structure (PCSS) descriptors related to the outer membrane, a conditional variational autoencoder (GenMTAMP) model was developed for de novo MTAMP design. Then, target PCSS descriptors within a predefined range are input as conditional constraints into the GenMTAMP model to direct generate MTAMPs. Candidate MTAMPs were screened and evaluated through subsequent identification (ClaAMP) and prediction (PreAMP) modules. Experimental validation showed that two top-ranked peptides named MTAMP003 and MTAMP004, exhibit excellent antibacterial activity against Gram-negative bacteria while maintaining low cytotoxicity and haemolytic activity toward mammalian cells. Furthermore, mechanism research indicates MTAMPs can disrupt the Gram-negative bacterial outer membrane barrier, with limited impact on mammalian cell membranes. In summary, the current research establishes a targeted and efficient generative artificial intelligence (AI) framework for de novo MTAMP design, and provides a generalisable framework for the rational design of AMPs with predefined functional properties.
|
|
Scooped by
mhryu@live.com
Today, 2:12 PM
|
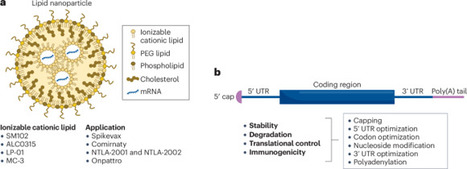
mRNA technology, which has enabled the rapid development of vaccines for infectious diseases, also holds great promise for a new generation of therapies for a host of rare and common diseases. A decade of clinical trials are beginning to clarify the key barriers to unlock the transformative potential of mRNA drugs, which are being addressed with novel interdisciplinary technical advances that, in some cases, are integrating the fields of gene, cell and mRNA therapies. Here, we review the scientific insights from a select group of clinical studies on mRNA-based drugs, including enzyme replacement therapies for rare diseases, cancer immunotherapies, genome-modifying therapies, and immune cell reprogramming therapies for cancer and autoimmune diseases. Several innovative approaches such as clinically tractable in vivo delivery systems, the development of completely ‘immune-silent’ mRNA–vehicle formulations that allow repeated administration and the development of approaches for preferential delivery to organs other than the liver would expedite the development of mRNA therapeutics 2.0. mRNA technology is being applied to develop a new generation of therapies for rare and common diseases. This Review discusses insights from clinical studies on mRNA-based therapeutics, including enzyme replacement therapies, cancer immunotherapies, genome-modifying therapies and immune cell reprogramming therapies, which illustrate how technological advances are paving the way for the wider adoption of mRNA technology in drug development.
|
|
Scooped by
mhryu@live.com
Today, 1:57 PM
|
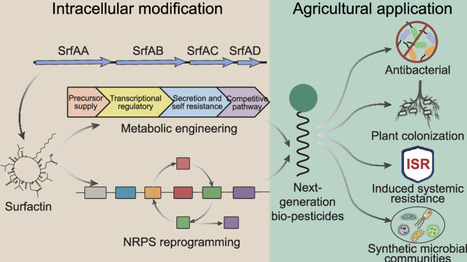
Surfactin, a cyclic lipopeptide produced by Bacillus species, represents a promising bio-pesticide due to its antimicrobial activity, environmental compatibility, and ability to induce plant immunity. However, fragmented knowledge on its molecular mechanisms and engineering limits the rational development of next-generation biopesticides. This review outlines surfactin biosynthetic principles and advances in metabolic and synthetic engineering strategies for improving yield and structural diversity. We emphasize high-throughput NRPS reprogramming strategies enabling precise control over peptide sequence and fatty acid composition, providing a powerful platform for the rational design of functionally tailored variants. Furthermore, surfactin may serve as an important signaling hub in the interaction network between plants and microorganisms. By integrating NRPS engineering, system metabolic design, and the construction of synthetic microbial communities, the future of crop protection is expected to shift from a single active molecule application model to a programmable ecological regulation model.
|
|
Scooped by
mhryu@live.com
Today, 1:39 PM
|
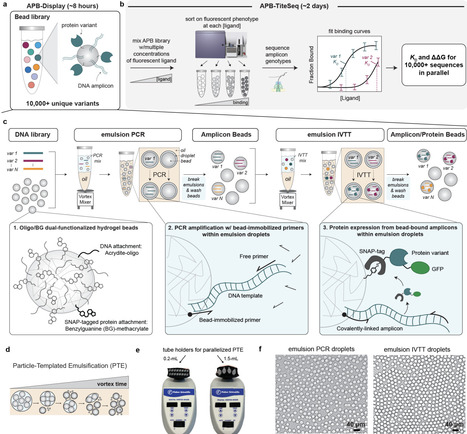
Scalable methods for producing and characterizing protein libraries are essential for generating standardized datasets needed to train models linking sequence to function. Here, we present Amplicon/Protein Bead Display (APB-Display), which expresses and purifies >100,000 protein variants in vitro in 1 day. APB-Display uses particle-templated emulsification to generate libraries of hydrogel beads that covalently display many copies of a given protein variant and its encoding DNA. By incubating beads with fluorescently-labeled ligands, sorting, and sequencing to generate titration curves (APB-TiteSeq), we simultaneously quantified expression levels and binding affinities (Kds) for >18,000 FLAG epitope variants interacting with M2 anti-FLAG antibody in 3 days, revealing chemistry-dependent epistasis between positions 4 and 5. Single-concentration binding measurements (APB-SortSeq) paired with neural network denoising further returned quantitative affinities for >88,000 variants. APB-Display requires only standard laboratory equipment and access to a FACS sorter, providing an accessible platform for quantitative in vitro biochemistry at scale.
|
|
Scooped by
mhryu@live.com
Today, 1:58 AM
|
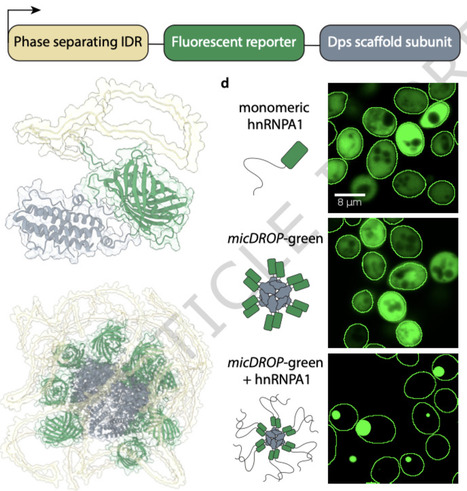
Intrinsically disordered regions (IDRs) drive intracellular phase separation and biomolecular condensate formation through interactions encoded in their sequence. Although condensates form spatially distinct assemblies in cells, the high conformational flexibility of IDRs and absence of well-defined 3D structures raise the question of how they could encode condensate specificity. To systematically characterize IDR–IDR interactions and their ability to mediate self-specific partitioning, we develop micDROP, a synthetic system of multivalent IDRs forming constitutive droplets. We examine ten natural IDRs that phase-separate in micDROP and find that their saturation concentrations in vivo correlate with total sequence stickiness. Co-expression of IDR pairs fused to distinct micDROP scaffolds reveals widespread promiscuity, whereas TDP43 and UBQ2 consistently form self-specific condensates. A short, conserved α-helical segment in the TDP43 IDR governs this self-recognition. Our results indicate that IDRs tune phase separation propensity through sequence composition, while discrete condensate identity likely requires additional structural determinants. Here the authors develop micDROP to systematically test IDR-IDR interactions driving condensate formation. Most IDR pairs co-mix promiscuously; only TDP43 and UBQ2 form distinct droplets, with a short α-helix in TDP43 driving self-recognition.
|
|
Scooped by
mhryu@live.com
Today, 1:51 AM
|
Genomovar-level and intragenomic diversity cannot be resolved by conventional amplicon sequencing due to the limitation of fragment lengths and read accuracy, while the application of metagenomic profiling to a large number of samples can be resource intensive. Here, we report UltraRes-rrn, an integrated wet-lab and computational workflow for high-accuracy rrn (i.e., 16S-ITS-23S rRNA) operon profiling using Nanopore sequencing. By integrating ultra-long DNA recovery, long-read amplicon sequencing, and unique molecular identifiers (UMI)-based consensus correction, UltraRes-rrn obtains full-length 16S-ITS-23S rRNA operon consensus sequences with mean accuracies exceeding 99.98%. To achieve higher resolution, we propose a hierarchical rRNA operon profiling strategy in which concatenated 16S+23S rRNA genes (16S23S) serve as a primary and the internal transcribed spacer (ITS) provides a secondary marker. The 16S23S marker achieved discrimination at the genomovar level compared to either 16S or 23S rRNA genes alone, which mitigates the ITS-driven over-splitting observed with the full-length rrn operon and allows for larger proportion of data being classified at higher confidence thresholds. Further, ITS variation was strongly structured by tRNA occurrence patterns, suggesting that ITS can capture taxon microdiversity missed by either 16S or 23S rRNA gene sequences alone. The UltraRes-rrn workflow was applied to full-scale nitrogen removal reactors, revealing intraspecies diversity variation driven by different carbon regimes, which would not have been possible with a shorter gene sequence. In summary, UltraRes-rrn enables cost-effective community profiling at genomovar-level resolution in complex ecosystems.
|
|
Scooped by
mhryu@live.com
Today, 12:59 AM
|
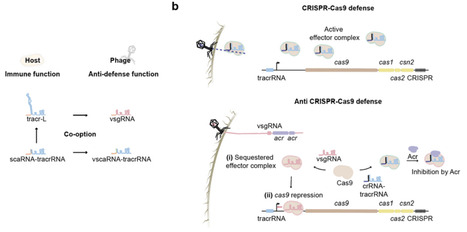
Central to CRISPR technologies is the single-guide RNA (sgRNA), an engineered fusion of a processed CRISPR RNA and tracrRNA that directs Cas9 and many Cas12 nucleases to bind and cleave target DNA. Here, we report the discovery of similarly compact viral sgRNAs (vsgRNAs) encoded by bacteriophages that counteract bacterial CRISPR-Cas9 immunity. vsgRNAs inhibit Cas9 function via two complementary routes: by sequestering the Cas9 apoenzyme, and by re-directing the Cas9 nuclease to transcriptionally silence its own promoter. vsgRNAs also can cooperate with co-encoded anti-CRISPR proteins (Acrs), including AcrIIA25.1 that blocks DNA binding by Cas9 complexed with a standard sgRNA but not with the vsgRNA. We predict that phages evolved vsgRNAs by co-opting and repurposing host-encoded long-form tracrRNAs (tracr-L) responsible for Cas9 auto-repression and a countermeasure to Acrs. Our search also uncovered Cas9-regulating small CRISPR-associated RNAs (scaRNAs), which we predict were inserted upstream of tracrRNAs to form tracr-L but were also co-opted by phages as viral scaRNAs to suppress Cas9 immunity. Finally, we found that vsgRNAs can enable genome editing in mammalian cells, offering a natural guide RNA template for CRISPR technologies. Overall, these findings reveal that bacteriophages devised their own compact sgRNAs tailored to subvert Cas9 immunity, long preceding their rational design to program RNA-guided nucleases.
|
|
Scooped by
mhryu@live.com
Today, 12:37 AM
|
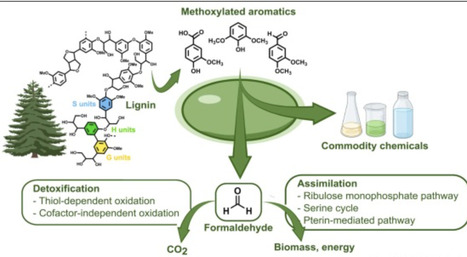
Formaldehyde is a toxic metabolite generated in the bacterial catabolism of lignin-derived aromatic compounds (LDACs) through the oxidative O-demethylation of methoxylated aromatics. Microbial cell factories designed to convert LDACs to commodity chemicals should ideally utilize formaldehyde to improve carbon efficiency and strain fitness. Methylotrophs and natural degraders of LDACs have evolved various strategies for utilizing formaldehyde. These range from cofactor-dependent detoxification systems that extract reducing equivalents to cyclical pathways that synthesize building blocks. In addition, recent engineering efforts, driven in part by interest in C1 feedstocks, have yielded useful synthetic formaldehyde metabolic pathways. In this review, we outline several of these routes, highlighting gaps and metabolic engineering opportunities, with a particular focus on improving LDAC biocatalysts.
|
|
Scooped by
mhryu@live.com
Today, 12:23 AM
|
The ever-expanding catalogue of uncharacterized proteins - the so called functional dark matter - poses a major challenge for biotechnological and biomedical exploitation. Functional assessment of most proteins is hindered by the technical limitations of annotation transfer and by the propagation of erroneous annotations in databases. The common denominator here is the reliance on sequence similarities. However, these become inaccurate below certain thresholds and can diverge even at sequence identities around 70%. To approach this challenge, we implemented a strategy using embeddings generated by protein language models for targeted function discovery (PE-TFD). Datasets of proteins representing target as well as non-target functions were used to train supervised learning models. The resulting ensemble models yielded interpretable prediction scores, enabling the exploration of databases without relying on multiple sequence alignments or structural information. We here tested PE-TFD for the discovery of novel hydrogenases as proof-of-concept, resulting in the novel discovery of 773 [NiFe] and 1,929 [FeFe] hydrogenases that were not detected by established sequence- or profile-based approaches. Structural analyses supported their non-random nature and further revealed a significant number of enzymes lacking prior functional annotation. Our framework therefore enables interpretable function discovery in large-scale datasets and the exploitation of functional dark matter.
|
|
Scooped by
mhryu@live.com
June 2, 11:32 PM
|
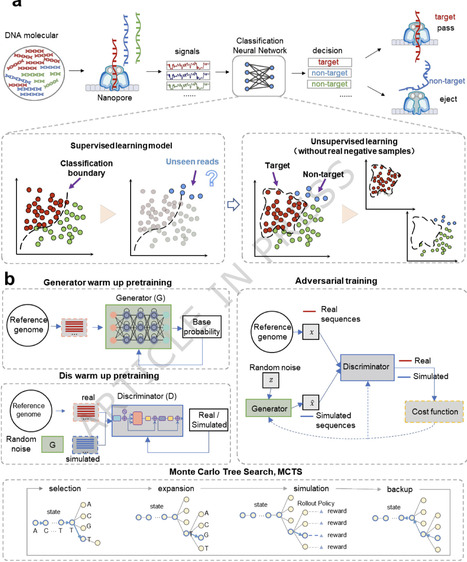
Nanopore adaptive sequencing enables real-time target enrichment, yet current deep-learning methods require costly, sample-specific experimental training data. To address this, we developed GANBase, a genome-guided generative adversarial learning framework, which is trained exclusively on reference sequences and incorporates a Monte Carlo Tree Search-based Rollout strategy for model training. GANBase demonstrates robust performance in target enrichment and host depletion across diverse scenarios. In live adaptive sequencing experiments, it remains effective despite significant pore loss or flow cell version updates, providing a data-independent solution that significantly expands the utility of real-time targeted sequencing. Adaptive nanopore sequencing enables targeted enrichment, but current methods rely on training data. Here, the authors develop GANBase, a genome‑guided deep‑learning framework that achieves robust, data‑independent target enrichment and host depletion across diverse sequencing conditions.
|
|
Scooped by
mhryu@live.com
June 2, 11:02 PM
|
Non-conventional yeasts offer unique advantages as tailored cell factories. Pichia pastoris is a master of protein expression and secretion. However, engineering P. pastoris for the overproduction of secondary metabolites is challenging due to its tightly regulated metabolism. How can we overcome this metabolic rigidity for efficient biosynthesis of energy-demanding molecules such as sclareol, a valuable diterpenoid? In this research article, we developed a host-specific framework integrating systematic pathway-level rewiring of the sclareol biosynthetic pathway and central metabolism to alleviate precursor acetyl-coenzyme A and cofactor NADPH limitations. Regulatory-level remodeling identified metabolic factors that enhance cellular robustness and protein homeostasis, reprogrammed global transcriptional regulators for carbon and nitrogen flux balance, and tailored process-level fermentation to eliminate byproducts. These efforts enabled a sclareol titer of 27.8 g l−1 in minimal medium, representing the highest diterpenoid production reported. This work redefines P. pastoris as a powerful chassis for natural product biosynthesis, providing a generalizable roadmap for engineering non-conventional hosts.
|
|
|
Scooped by
mhryu@live.com
Today, 2:34 PM
|
The microbiota produces thousands of potentially bioactive small molecules. High-throughput bioactivity screens of in vitro commensal cultures have exposed microbiota metabolites that shape host physiology by activating diverse G-protein-coupled receptors (GPCRs). However, owing to technical limitations, the GPCRome-wide bioactivities of in vivo metabolomes, which result from complex diet–microorganism–host interactions, remain unclear. Here we used a multiplexed GPCR screening technology to assess GPCRome-wide bioactivities of 100 commensal strains grown in vivo in monoassociated germ-free mice or in vitro in bacterial culture medium. In vivo and in vitro commensal metabolomes exhibited distinct GPCR activation patterns due to (1) host-mediated metabolite degradation; (2) in vivo microbial metabolic reprogramming; and (3) biotransformation of dietary substrates. Notably, we found that multiple commensal strains produced acetylcholine (ACh) in vivo through the conversion of dietary choline, including select Bifidobacterium strains that dominate the microbiome in early life and a probiotic Pediococcus strain. Mechanistically, we identified and characterized the bacterial enzymes that mediate this biotransformation in Bifidobacterium breve and Pediococcus pentosaceus, and generated an isogenic mutant B. breve strain lacking ACh production. Mice colonized with ACh-producing B. breve exhibited enhanced intestinal immunoglobulin A (IgA) production, altered microbiota composition and increased resistance to enteric infection. These findings underscore the profound impacts of the in vivo environment on microbiota metabolism and reveal a diet–microbiome–host axis that strengthens mucosal immune defences and reinforces host–microbiota mutualism. A diet–microbiome–host axis strengthens mucosal immune defences and reinforces host–microbiota mutualism.
|
|
Scooped by
mhryu@live.com
Today, 2:26 PM
|
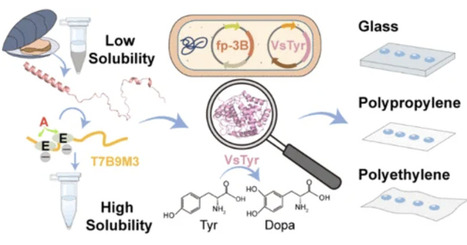
Due to intrinsically low solubility and stability, recombinant mussel foot proteins (Mfps) require an efficient system that supports large-scale production of dopanized-Mfps. Here, we engineered a 39-aa peptide tag, termed T7B9M3, to increase the solubility of Mfps in E. coli. The absence of net charged residues, rather than the copy number or the predicted α-helical structure of the T7B9M3 tag, contributes to the solubility enhancement. T7B9M3-fp-3B achieved a 365.96 mg/L soluble protein titer with a 98.75% soluble rate in a shake flask. Coupling the tyrosinase coexpression to endow Mfps with a dopaminization capability, the tyrosine hydroxylation efficiency reached 19.82%. Moreover, dopanized-Mfps tended to form stronger interactions with various substrates, including glass, polypropylene, and polyethylene, than the unmodified version. Our findings suggest that the combined strategy may contribute to establishing a scalable and low-cost protein expression platform with the potential for batch production of functional Mfps.
|
|
Scooped by
mhryu@live.com
Today, 2:15 PM
|
Current long-read single-nucleotide variant callers were designed primarily for genomic data—particularly human genomes. While some have been used on metagenomic data, their underlying assumptions and training procedures fail to account for the inherent complexity of metagenomic samples. To date, no long-read variant caller has been purpose-built for metagenomic applications. To address this gap, we present SNooPy, a single nucleotide polymorphism (SNP)-calling tool that implements a new statistical framework tailored to long-read metagenomic data. Unlike previous genomic methods, our approach makes no assumptions about the number of haplotypes present, their evolutionary relationships, or their sequence divergence. We demonstrate that SNooPy outperforms both traditional statistical and deep learning–based SNP callers. Our results suggest that future integration of this framework with deep learning approaches could further enhance variant-calling performance. SNooPy is freely available on github.com/rolandfaure/snoopy.
|
|
Scooped by
mhryu@live.com
Today, 2:00 PM
|
5-Aminolevulinic acid (5-ALA) is a naturally occurring nonproteinogenic amino acid with considerable potential in agricultural and pharmaceutical applications, and its biomanufacturing has attracted growing interest. To meet market demand, this study developed an efficient production process by engineering E. coli. First, we established the C4 biosynthetic pathway for 5-ALA and minimized glycine degradation by targeted deletion of gcvP and kbL. Subsequently, the introduction of a nonoxidative glycolysis (NOG) module combined with enhanced glucose uptake further increased 5-ALA production. To alleviate host toxicity, efflux engineering and antioxidant systems were integrated, further raising the titer to 6.93 ± 0.28 g/L. Dynamic regulation of sucCD expression together with redox rebalancing maintained a balance between precursor supply and cell growth. Finally, attenuation of hemB expression reduced metabolic flux diversion toward heme synthesis, yielding the engineered strain A48, which produced 55.11 g/L of 5-ALA within 41 h in a 5 L bioreactor. The metabolic engineering strategy presented in this study establishes an efficient biosynthetic platform for 5-ALA production.
|
|
Scooped by
mhryu@live.com
Today, 1:55 PM
|
Heparin has been the most important drug for treating thrombotic disorders for more than 60 years. However, the traditional production of heparin involves the slaughter of animals. Therefore, there is a demand for the animal-free production of heparin, such as enzymatic synthesis based on the heparin biosynthetic pathway. To achieve this, robust 6-O-sulfotransferases (6OSTs) are required to produce the 6-O-sulfation pattern in heparin, which is crucial for the biological activity. However, most native 6OSTs are derived from animal tissues and exhibit poor recombinant expression, low catalytic efficiency, and insufficient stability in E. coli. To overcome these limitations, we systematically established a two-tier engineering framework that integrated structure-guided protein repair and optimization of translation. Cross-species screening identified Oryzias melastigma 6OST-1 as an engineering-competent template. First, we constructed the variant 6OST-M10 through protein repair one-stop service-guided structural restoration combined with targeted reverse mutations. To address the long-standing challenge of low heterologous expression, we generated a synonymous rare-codon (SRC)-guided ultrahigh-throughput screening platform based on split-GFP complementation. This platform systematically tunes the translation kinetics for sulfotransferases. Ultimately, the 6OST-M10(SRC) variant was generated, which displayed an 8.375-fold increase in soluble expression and a 27-fold improvement in catalytic activity that reached 4400 IU/L under high-density fermentation. This dual-layer strategy couples structural stabilization with translational optimization to resolve the trade-offs among activity, stability, and recombinant expression that have previously limited bacterial production of animal-derived sulfotransferases and heparin synthesis in E. coli. This framework provides a universal, scalable paradigm for protein engineering and enables the synthesis of bioengineered heparin and other glycosaminoglycan products without the involvement of animals.
|
|
Scooped by
mhryu@live.com
Today, 2:02 AM
|
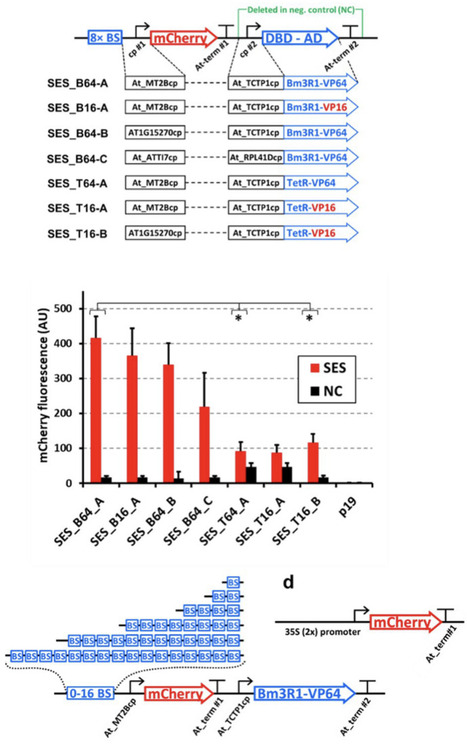
Plants represent commercially relevant production systems for recombinant proteins and chemical compounds. Effective genetic engineering depends on precise control of heterologous gene expression, which remains challenging due to complex transcriptional and post-transcriptional regulation, endogenous gene silencing mechanisms, and notably because of limited number of tools for allowing robust, fine-tuned control of expression levels across different systems/organisms. Some of the most common issues associated with plant expression systems are addressed with our plant-optimized version of a previously developed fungal universal synthetic expression system (SES). Plant SES demonstrates several favorable characteristics for robust heterologous gene expression, including highly constitutive function with apparently reduced sensitivity to endogenous silencing in transient assays, without requiring p19 co‑expression under the tested conditions. These together provide simple, predictable tuning of gene expression levels, and the potential for very high expression levels of the target genes. In all these features, SES shows higher and more stable transcript levels than Cauliflower Mosaic Virus (CaMV) 35 S promoter-based constructs in our experimental setups. The functionality of plant SES was tested by expressing mCherry and three commercially relevant proteins: fungal glucose oxidase (GOX), protein A and human vascular endothelial growth factor 165 (VEGF16) from diverse organisms, supporting high-level accumulation of recombinant proteins. In addition, plant SES retains full functionality in both plant and fungal hosts, which makes this expression system a useful tool for a multitude of genetic engineering applications in other eukaryotic organisms.
|
|
Scooped by
mhryu@live.com
Today, 1:53 AM
|
Plasmids benefit bacterial communities by storing auxiliary genes that address environmental challenges such as antibiotics. Subsequent plasmid loss can also be advantageous if plasmid benefits are temporary but costs are permanent. However, unless positive selection is sustained, plasmid loss can proceed to extinction, with access to plasmid-derived benefits permanently lost. In principle, horizontal transmission can maintain a plasmid in a population, but if the plasmid cost is too high, the host can become uncompetitive. We examine how survival of costly but occasionally beneficial plasmids is possible in a bacterial population. Using population models, we demonstrate that plasmid-dependent phages can, counterintuitively, solve this plasmid survival problem for their bacterial hosts. Phage predation pins the plasmid at low but nonzero abundance, such that the plasmid cost is effectively neutralized at the population level, dramatically lengthening the persistence time of the plasmid. When conditions change and the costly plasmid becomes beneficial, it spreads across the host population and switches to a vertical-transmission lifestyle until benefits again subside.
|
|
Scooped by
mhryu@live.com
Today, 1:25 AM
|
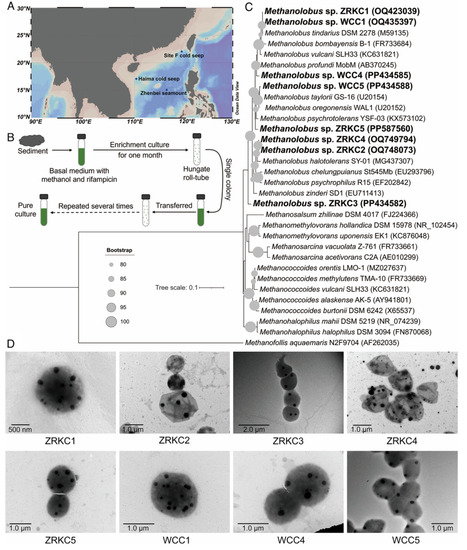
Microorganisms employ inorganic polyphosphate (polyP) as an ancient strategy for energy and phosphate storage, yet its physiological roles in methanogenic archaea remain largely unexplored. Here, we report that polyP metabolism is coupled to growth and methanogenesis in Methanolobus sp. ZRKC1, a representative of eight methanogenic archaea isolated from deep-sea cold seep sediments. Through combined genetic, biochemical, and physiological analyses, we find that PPK1 mediates polyP synthesis in a Mg2+-dependent manner, whereas PPK2 functions primarily as a polyP hydrolase. Deletion of ppk1 in strain ZRKC1 abolishes polyP accumulation and impairs both growth and methane production, pointing to a role for polyP as a metabolic hub linking these processes. Transcriptomic profiling reveals that under organic phosphorus conditions, strain ZRKC1 upregulates phosphoesterases to liberate bioavailable phosphate, which is subsequently channeled into polyP via PPK1. Furthermore, in situ transcriptomic data suggest that the genetic capacity for this metabolic strategy may be present and transcriptionally active in the native environment, with concurrent upregulation of genes involved in phosphate acquisition, polyP metabolism, and methanogenesis. Our findings suggest the importance of polyP in linking phosphate homeostasis to growth and methanogenesis in deep-sea methanogenic archaea.
|
|
Scooped by
mhryu@live.com
Today, 12:45 AM
|
Studies of whole viral populations—the “virome”—are yielding exciting new insights into biological systems, but methods are still being optimized. Here, we describe generation and use of a synthetic viral community and its use to evaluate technical challenges arising in virome analysis. We spiked the mock community into different human sample types, then passed the samples through different virus enrichment protocols and analyzed by Illumina sequencing. Compared with direct metagenomic sequencing, VLP enrichment protocols greatly increased viral read yields from stool and saliva. Four methods for DNA amplification were compared, with three showing over-amplification of small circular ssDNA viruses, most notably GenomiPhi. Studies of viral particle stability in the presence of nuclease showed that most viral genomes were stable when protected in viral particles, but phage MS2 RNA was unexpectedly labile under some of the conditions tested. Comparison of Illumina 1,000-cycle sequencing versus 300-cycle sequencing showed that longer reads supported generation of longer viral genome assemblies. We tested bacteriophage T4 DNA modified with glucosyl-hydroxymethylcytosine (ghmC) and hydroxymethylcytosine (hmC) and found that both were readily detected, though the recovery of ghmC-modified DNA was reduced compared with T4 genomes with unmodified cytosine. These studies together with published data help provide guidance for virome researchers optimizing analytical protocols.
|
|
Scooped by
mhryu@live.com
Today, 12:34 AM
|
Connecting mathematical models with empirically measured microbial growth has remained challenging, as numerous competing models based on different theoretical approaches can fit observations. Therefore, we develop a method to automatically propose growth models from microbial data alone. We validate this approach using an available dataset of E. coli grown on known resources, and study 14 species across various concentrations of a rich medium. The inherently interpretable approach of symbolic regression infers explicit dynamical models directly from growth data. Using symbolic regression natively, does not favor biologically interpretable models, but we find cumulative population gain to be a more informative machine learning feature than population size. Random Forest machine learning allows us to relate this finding to the approximation of a constant-rate per capita resource consumption. This suggests that the area under the growth curve (AUC) measured in routine experiments provides information on the effective resource dynamics governing microbial growth. Finally, we use theoretical insights to inform the symbolic regression algorithm and favor biologically interpretable models. Overall, we found that balancing between data fit, parsimony and biological relevance favored both the simplest, linear approximation and models based on Monod dynamics, with either one or two underlying resources. Therefore, our approach to read growth laws off of microbial batch cultures provides insights on data-driven modelling.
|
|
Scooped by
mhryu@live.com
Today, 12:04 AM
|
Environmental samples contain complex microbial communities hiding a treasure trove of active compounds. However, screening for active natural products from environmental samples is challenging due to inefficient cultivation techniques and a lack of proper screening platforms. For empowering antibiotic screening assays from complex microbial communities, we have developed and optimized a droplet-based platform with multiplexing capability. A cultivation strategy for bacteria in picoliter droplets was combined with phenotypic screening using multiple whole-cell reporter species. A mixture of two different fluorescently labelled reporter strains, one Gram-positive and one Gram-negative, was picoinjected to each of millions of picoliter cultures, which were screened for inhibiting activity based on the independent survival signals of each reporter species. Proof-of-concept experiments demonstrate efficient detection, selection, and recovery of a model Streptomyces strain from a synthetic mixture according to the specific inhibition of the reporter strains by the produced antibiotic. Subsequently, the established platform was successfully applied to screen environmental microbial communities from soil samples. This approach showcases multiplexing capabilities for screening assays in microfluidic droplets in order to simultaneously screen for new bioactive compounds with various inhibition profiles.
|
|
Scooped by
mhryu@live.com
June 2, 11:27 PM
|
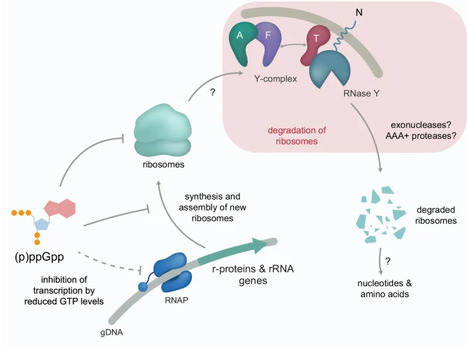
Limiting ribosome synthesis and activity is crucial for adaptation to stresses, such as heat or nutrient starvation. In Bacillus subtilis, this can be achieved through the coordinated action of the alarmones (p)ppGpp and the transcription factor Spx. Here, we performed a genetic screen to identify novel factors that contribute to the heat shock response in B. subtilis. We identified the Y-complex, which confers specificity to the endonuclease RNase Y, as a critical player under stress conditions, such as heat or transition into the stationary phase. This protein complex is required for the targeting and processing of diverse RNAs, notably the maturation of mRNAs encoding proteins involved in translation and metabolism. We further demonstrate that the Y-complex and RNase Y initiate the degradation of rRNAs of mature ribosomes, lowering their abundance. We propose that the Y-complex is a regulatory hub that modulates gene expression, adjusts protein synthesis and resource allocation. Transcriptional regulation is a central mechanism underlying bacterial adaptation to stress. Here, the authors used a genetic fitness screen to show that the Y-complex in Bacillus subtilis contributes to the heat shock response through its interaction with the ribosome.
|























program finds the best mathematical equation describing how that organism grows