 Your new post is loading...

|
Scooped by
?
Today, 4:37 PM
|
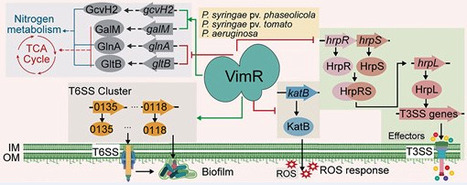
Pseudomonas syringae, a highly destructive plant bacterial pathogen causing severe disease and significant yield losses in agriculture globally, has complex regulatory systems involving many transcriptional factors (TFs). Although the LysR-type transcriptional regulator (LTTR) protein family is a well-known group of TFs involved in diverse physiological functions, the roles of LTTRs in P. syringae remain largely unknown. In this study, we characterized a LysR-type TF, PSPPH4638, and designated it as the virulence and metabolism regulator VimR. Genome-wide identification of VimR using chromatin immunoprecipitation sequencing revealed 1032 binding sites in the genome, of which 85% were in intergenic regions. Transcriptomic analysis showed altered expression of 454 and 82 genes in response to ΔvimR in King’s B medium (KB) and minimal medium (MM), respectively. Conjoint analysis showed that 99 genes were directly affected by VimR in KB. VimR was identified as a repressor of the type III secretion system, oxidative stress response, and key metabolic pathways such as the tricarboxylic acid cycle. In addition, we found that VimR was positively involved in the type VI secretion system and alanine, aspartate, and glutamate metabolism. Further verification showed that VimR was widely present in Pseudomonas, displaying similar binding capacity in different strains of P. syringae, and similar regulatory functions in Pseudomonas aeruginosa. Taken together, our findings identified a conserved master TF that regulates type III secretion system, type VI secretion system, and multiple metabolic pathways in Pseudomonas.
|
|
Scooped by
?
Today, 4:20 PM
|
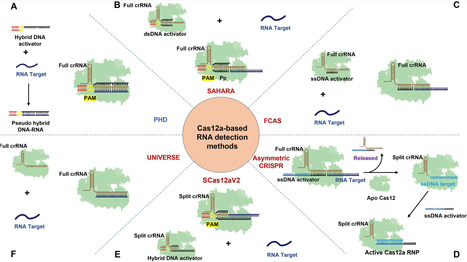
The CRISPR/Cas12a system is known for its intrinsic RNA-guided trans-cleavage activity; however, its RNA detection sensitivity is limited, with conventional methods typically achieving detection limits in the nanomolar range. Here, we report the development of a “pseudo hybrid DNA–RNA” (PHD) assay that significantly enhances the RNA detection capability of Cas12a. The PHD assay achieves a striking detection limit of 7.7 pM using single CRISPR RNA (crRNA) and 33.8 fM using pooled crRNAs. Importantly, this assay exhibits ultra-high specificity, capable of distinguishing mutated RNA target sequences at the protospacer adjacent motif (PAM)-distal region. It can also detect ultrashort RNA sequences as short as 6–8 nt and long RNAs with complex secondary structures. Additionally, the PHD assay enables PAM-free attomolar-level DNA detection. We further demonstrate the practical utility of the PHD assay by successfully detecting miR-155 biomarkers and human pappilloma virus 16 DNA in clinical samples. We anticipate that the design principles established in this study can be extended to other CRISPR/Cas enzymes, thereby accelerating the development of powerful nucleic acid testing tools for various applications.
|
|
Scooped by
?
Today, 4:16 PM
|
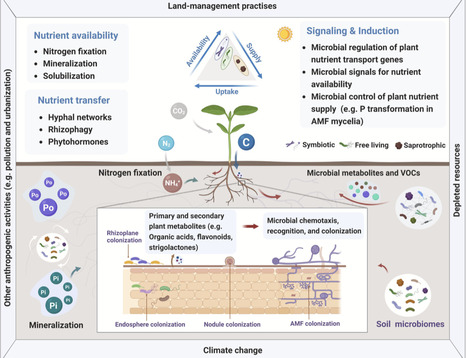
Plants and microbiomes have co-evolved for millennia. Through this co-evolution, microbiomes have become essential for plant nutrient acquisition, which involves plant signaling, microbial sensing, and acquiring and sharing nutrients. In this review, we synthesize recent advancements in the complex associations of molecular, physiological, and eco-evolutionary mechanisms that underpin microbe-facilitated plant nutrient uptake. Focusing on emerging insights in plant-microbial communication and metabolic pathways, we evaluate potential opportunities to harness plant microbiomes to sustainably supply nutrients in agricultural and natural ecosystems. However, further progress is constrained by key knowledge gaps. We propose an amended conceptual framework for advancement that includes a holistic understanding of eco-evolutionary relationships with explicit consideration of signaling and sensing mechanisms. Finally, we argue that advancing fundamental science by utilizing emerging analytical approaches in an integrated way is critical to develop effective microbiome-informed tools that can enhance plant nutrient acquisition and promote long-term food security and environmental sustainability.
|
|
Scooped by
?
Today, 3:57 PM
|
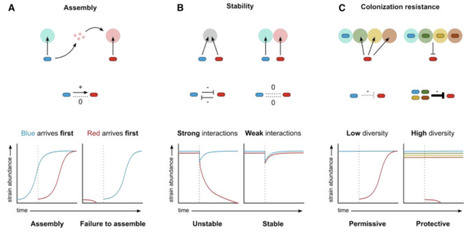
Many plants and animals, including humans, host diverse communities of microbes that provide many benefits. A key challenge in understanding microbiomes is that the species composition often differs among individuals, which can thwart generalization. Here, we argue that the key to identifying general principles for microbiome science lies in microbial metabolism. In the human microbiome and in other systems, every microbial species must find ways to harvest nutrients to thrive. The available nutrients in a microbiome interact with microbial metabolism to define which species have the potential to persist in a host. The resulting nutrient competition shapes other mechanisms, including bacterial warfare and cross-feeding, to define microbiome composition and properties. We discuss impacts on ecological stability, colonization resistance, nutrient provision for the host, and evolution. A focus on the metabolic ecology of microbiomes offers a powerful way to understand and engineer microbiomes in health, agriculture, and the environment.
|
|
Scooped by
?
Today, 3:36 PM
|
Agricultural long-term field trials provide fundamental data on crop performance and soil characteristics under diverse management practices. This information represents essential knowledge for upcoming challenges in food and nutrition security. Data provided here have been compiled since 2004 from a nitrogen(N)-fertilization intensity, tillage, and crop rotation field trial in Central Germany including standardized metrics regarding soil management, physical soil properties, crop management, crop characteristics, yield, and harvest quality parameters. In 2015, the field trial became a member of the German Agricultural Soil Research Program BonaRes. Numerous measurement results were added including plant physiology and soil and rhizosphere microbiology. DNA of bacterial/archaeal and fungal microbiomes was sequenced in the rhizosphere and root-associated soil following a meta-barcoding approach. Taxonomic and relative abundance data were included in the dataset. The dataset is the first to include information on root characteristics, soil and rhizosphere microbiomes, and crop gene expression. We encourage reuse of these biological field trial data in terms of meta-analysis, modeling and AI approaches.
|
|
Scooped by
?
Today, 12:38 AM
|
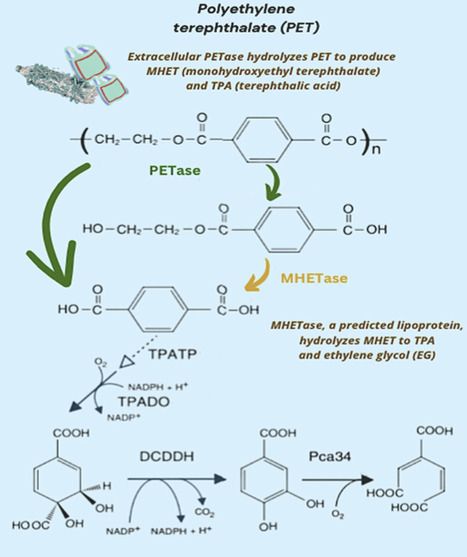
Additives such as stabilizers, plasticizers, and fillers are commonly used in relatively small amounts to enhance the structure of plastics. Notably, some of these additives, including moieties of compounds containing nitrogen, phosphorus, and sulfur, are essential for microbial proliferation. Most studies on plastic degradation have primarily focused on the potential of microorganisms to assimilate carbon from plastics to support their growth, a strategy that has yet to yield significant success. However, studies investigating the removal of non‑carbon moieties of additives from plastics, which could weaken their structure and thereby enhance fragmentation, remain largely unexplored. This review highlights the potential of harnessing microbial processes that target the non‑carbon moieties of additives to weaken the structural integrity of plastics. The weakened plastic may then become more accessible to heterotrophic microbes, thereby accelerating its degradation.
|
|
Scooped by
?
Today, 12:10 AM
|
Structural data provides important information on the proteins’ function. Recent development of advanced machine learning and artificial intelligence tools, such as AlphaFold, have led to an explosion of predicted protein structures. However, many of the computed protein models contain unstructured and disordered regions, posing challenges in protein function characterization. Here we present BindUP-Alpha, an upgraded webserver for predicting nucleic acid binding proteins. Our structure-based algorithm utilizes the electrostatic features of the protein surface and other physiochemical and structural properties extracted from the protein sequence. Using a Support Vector Machine (SVM) learning approach, BindUP-Alpha successfully predicts DNA- and RNA-binding proteins from both experimentally solved structures and predicted models. In addition, BindUP-Alpha identifies electrostatic patches on the protein’s surface that represent potential nucleic-acid binding interfaces. BindUP-Alpha is freely accessible at https://bindup.technion.ac.il, providing interactive three-dimensional visualizations and downloadable text-based results.
|
|
Scooped by
?
June 11, 11:37 PM
|
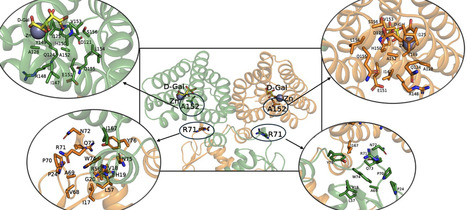
Genetic variations in transcriptional regulators (TRs) of metabolic loci can influence host-bacterial interactions by affecting carbon utilization. Although the metabolism of sugar acids, including D-galactonate, is extensively implicated in the colonization and virulence of enteric bacteria, there has been no investigation on the extent of variations in their pathway-specific TRs. DgoR, the TF of D-galactonate metabolism, is the best-characterized GntR/FadR family sugar acid TFs in enteric bacteria, recognized by the presence of an N-terminal winged helix-turn-helix DNA-binding domain and a C-terminal effector-binding and oligomerization (E-O) domain connected by a linker. Here, we examined 340 Escherichia coli isolates for variations in dgoR and studied their effect on repressor function. Genetic and biochemical studies identified variants with a partial loss of DNA-binding ability (P24L and A152E) and a decreased response to D-galactonate (R71C and P92L). Because the linker residue R71C resulted in a reduced response to D-galactonate and the E-O domain residue A152E led to a DNA binding defect, we performed simulations to probe their altered allosteric behavior. We observed that the correlation patterns, dynamics, and networks of the variants are indeed distinct from the wild type. Importantly, corroborating their repressor function, R71C and A152E variations impacted the growth of natural isolates in D-galactonate. Alignment-based variation detection across all E. coli and Enterobacterales identical protein group data sets revealed less prevalence of these four variations. Collectively, the present study highlights the need for a thorough analysis of the effect of variations in sugar acid TRs on repressor function and their effect on host-bacterial interactions.
|
|
Scooped by
?
June 11, 11:07 PM
|
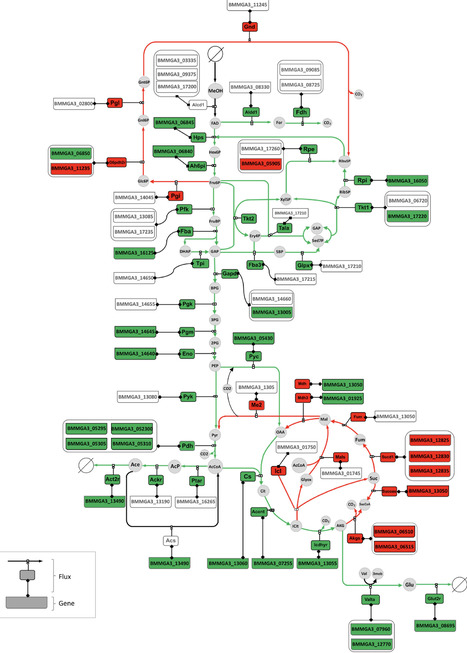
Bacillus methanolicus is the next workhorse in biotechnology using methanol, an alternative and economical one-carbon feedstock that can be obtained directly from carbon dioxide, as both carbon and energy source for the production of value-added chemicals. The wild-type strain B. methanolicus MGA3 naturally overproduces l-glutamate in methanol-based fed-batch fermentations. Here we generated a B. methanolicus strain exhibiting enhanced l-glutamate production capability through induced mutagenesis. To showcase the potential of this mutant strain, further metabolic engineering enabled the production of γ-aminobutyric acid (GABA) directly from l-glutamate during methanol fed-batch fermentations. Using a systems-level analysis, encompassing whole-genome sequencing, RNA sequencing, fluxome analysis and genome-scale metabolic modelling, we were able to elucidate the metabolic and regulatory adaptations that sustain the biosynthesis of these products. The metabolism of the mutant strain specifically evolved to prioritize energy conservation and efficient carbon utilization, culminating in increased product formation. These results and insights provide a foundation for further rational metabolic engineering and bioprocess optimization, enhancing the industrial viability of B. methanolicus for sustainable production of l-glutamate and its derivatives.
|
|
Scooped by
?
June 11, 7:42 PM
|
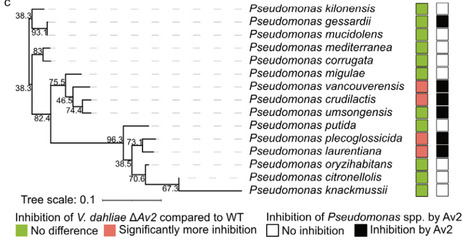
During pathogen attack, plants recruit beneficial microbes in a cry for help to mitigate disease development. Simultaneously, pathogens secrete effectors to promote host colonization through various mechanisms, including targeted host microbiota manipulation. Here, we characterize the Av2 effector of the vascular wilt fungus Verticillium dahliae as a suppressor of the cry for help. Inspired by in silico antimicrobial activity prediction, we investigated the antimicrobial activity of Av2 in vitro. Furthermore, its role in V. dahliae virulence was assessed through microbiota sequencing of inoculated plants, microbial co-cultivation assays, and inoculations in a gnotobiotic plant cultivation system. We show that Av2 inhibits bacterial growth, and acts as a virulence factor during host colonization. Microbiota sequencing revealed involvement of Av2 in suppression of Pseudomonas spp. recruitment upon plant inoculation with V. dahliae, suggesting that Av2 suppresses the cry for help. We show that several Pseudomonas spp. are antagonistic to V. dahliae and sensitive to Av2 treatment. We conclude that V. dahliae secretes Av2 to suppress the cry for help by inhibiting the recruitment of antagonistic Pseudomonas spp. to pave the way for successful plant invasion.
|
|
Scooped by
?
June 11, 7:10 PM
|
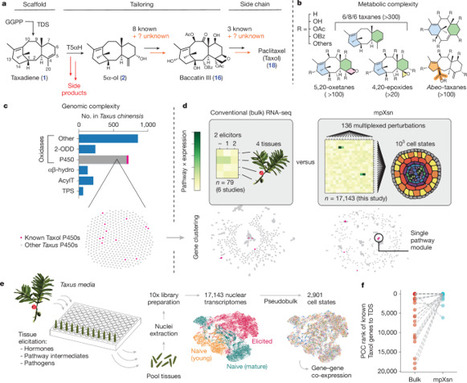
Plants make complex and potent therapeutic molecules, but sourcing these molecules from natural producers or through chemical synthesis is difficult, which limits their use in the clinic. A prominent example is the anti-cancer therapeutic paclitaxel (sold under the brand name Taxol), which is derived from yew trees (Taxus species). Identifying the full paclitaxel biosynthetic pathway would enable heterologous production of the drug, but this has yet to be achieved despite half a century of research. Within Taxus’ large, enzyme-rich genome, we suspected that the paclitaxel pathway would be difficult to resolve using conventional RNA-sequencing and co-expression analyses. Here, to improve the resolution of transcriptional analysis for pathway identification, we developed a strategy we term multiplexed perturbation × single nuclei (mpXsn) to transcriptionally profile cell states spanning tissues, cell types, developmental stages and elicitation conditions. Our data show that paclitaxel biosynthetic genes segregate into distinct expression modules that suggest consecutive subpathways. These modules resolved seven new genes, allowing a de novo 17-gene biosynthesis and isolation of baccatin III, the industrial precursor to Taxol, in Nicotiana benthamiana leaves, at levels comparable with the natural abundance in Taxus needles. Notably, we found that a nuclear transport factor 2 (NTF2)-like protein, FoTO1, is crucial for promoting the formation of the desired product during the first oxidation, resolving a long-standing bottleneck in paclitaxel pathway reconstitution. Together with a new β-phenylalanine-CoA ligase, the eight genes discovered here enable the de novo biosynthesis of 3’-N-debenzoyl-2’-deoxypaclitaxel. More broadly, we establish a generalizable approach to efficiently scale the power of co-expression analysis to match the complexity of large, uncharacterized genomes, facilitating the discovery of high-value gene sets. An approach that combines single-nucleus RNA sequencing and multiplexed perturbation identifies genes that enable the biosynthesis of direct precursors of the anti-cancer drug Taxol, whose current production involves a laborious extraction process from yew trees.
|
|
Scooped by
?
June 11, 6:37 PM
|
Phage therapy is emerging as a promising strategy against the growing threat of antimicrobial resistance, yet phage and bacteria are incredibly diverse and idiosyncratic in their interactions with one another. Clinical applications of phage therapy often rely on a process of manually screening collections of naturally occurring phages for activity against a specific clinical isolate of bacteria, a labor-intensive task that is not guaranteed to yield a phage with optimal activity against a particular isolate. Herein, we review recent advances in artificial intelligence (AI) approaches that are advancing the study of phage-host interactions in ways that might enable the design of more effective phage therapeutics. In light of concurrent advances in synthetic biology enabling rapid genetic manipulation of phages, we envision how these AI-derived insights could inform the genetic optimization of the next generation of synthetic phages.
|
|
Scooped by
?
June 11, 6:04 PM
|
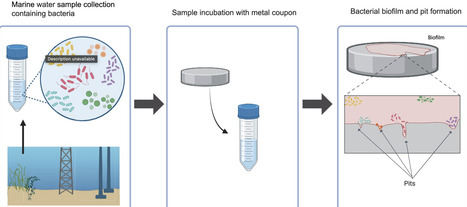
Steel corrosion is an extensive problem worldwide, substantially impacting marine infrastructures. In this study, the influence of steel on bacterial succession and corrosion was investigated by culturing marine water samples with and without steel coupons for 14 days. Compared to abiotic controls, oxygen levels were rapidly depleted in biotic cultures. Fe levels increased in controls compared to biotic cultures, potentially due to anoxic conditions and the incorporation of Fe in the biofilm. Proteobacteria dominated the initial cultures, but over 14 days the number of phylogenetic groups decreased overall in abundance. Taxons that increased in abundance included Clostridiaceae, Fusobacteriaceae, Flavobacteriaceae and Prolixibacteraceae, some members of which can utilize Fe. While initially in low abundance, Arcobacteraceae, Pseudoalteromonadaceae, Rhodobacteraceae and Rhizobiaceae numbers increased over time. Sites 1 and 2 cultures displayed localised deep pitting corrosion on coupon surfaces, consistent with microbial action, with an increase in Bacteroidetes, suggesting this phylum facilitates corrosion. In contrast, Site 3 cultures displayed uniform, superficial corrosion, with Clostridiaceae being the dominating family by Day 14, suggesting corrosion inhibition through biofilm formation. By identifying bacteria associated with corrosion, targeted approaches to corrosion reduction may be developed through identifying significant metabolic pathways by transcriptomics and the application of metabolic inhibitors.
|
|
|
Scooped by
?
Today, 4:33 PM
|
Functional analysis of bacterial genes or genomic fragments in vivo primarily relies on the analysis of knockout strains. Although various methods have successfully generated bacterial knockout mutants, the parallel operation of multiple sites, especially in biofoundries, remains challenging. New technological refinements of existing methods are necessary for high-throughput genomic editing in bacteria. In this study, to modify numerous sites in parallel, we optimized the linear donor DNA by adding modification at the different positions and achieved high-efficiency recombination with chemical transformation. Then, by combining with the CRISPR system, we established a guide sequence-independent and donor DNA-mediated genomic editing (GIDGE) method, enabling efficient and scarless engineering of common E. coli strains as well as wild-type strains such as E. coli MG1655, with particularly marked advantages demonstrated in E. coli Nissle 1917. This method allows for high-throughput genomic engineering in a 96-well format and is useful for sequence deletion with a wide range of lengths, sequence insertion, sequence replacement, and point mutation. As a proof-of-concept study, we constructed 96 single-gene knockout mutants and a genomic large-fragment deletion library in E. coli K-12 MG1655 using the GIDGE method. This high-throughput and easy-to-use method has great potential for automation and can be adapted for use in biofoundries.
|
|
Scooped by
?
Today, 4:17 PM
|
Using modern genomic tools, Feng et al. revisited Mendel’s seven pea traits in a recent Nature study, uncovering the molecular genetic basis of all of them, including the three unresolved ones: pod color, pod shape, and flower position. Their work highlights the level of complexity provided by structural variation that could impact genes and their regulatory regions and thus influence the expression of plant traits. The authors demonstrate how revisiting foundational experiments with contemporary tools can manifest novel biological insights and also guide future crop improvement.
|
|
Scooped by
?
Today, 4:02 PM
|
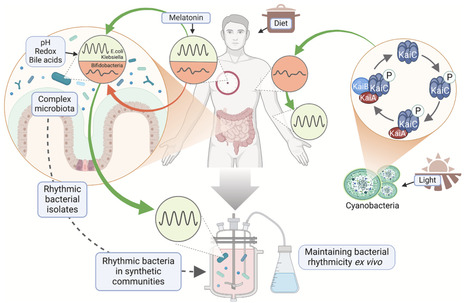
Daily oscillations in microbiota composition and function are emerging as an important element in host-microbiota interactions. Here, we summarize features of the microbiota that undergo diurnal rhythms, their development, their impact on the biology of the host, and their relevance to human health and disease. In particular, we focus on the intrinsic and extrinsic factors that regulate microbiota oscillations and the multifaceted roles that microbiota rhythmicity plays in host physiology, immunity, and metabolism. Given the pervasive impact of intestinal microorganisms on host health, understanding the origins and functions of microbiota rhythms is a critical aspect of the circadian biology of the meta-organism.
|
|
Scooped by
?
Today, 3:50 PM
|
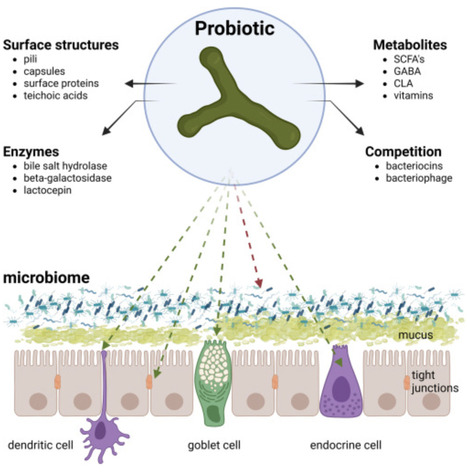
Probiotics, prebiotics, synbiotics, and postbiotics are required, by definition, to confer a health benefit on the host. It is often presumed the host microbiome plays a central role in the mechanism of action of these substances, collectively referred to here as “biotics.” However, the definitions of both probiotics and postbiotics do not include an associated mechanism nor the involvement of the microbiome. The definitions of prebiotics and synbiotics require evidence of selective utilization by the host microbiome, but do not state that confirmatory evidence must be provided that this utilization causes the associated health benefit. In this perspective, we discuss evidence supporting a role for the microbiome in delivering these health benefits and whether or not measuring microbiome alterations can serve as important readouts of efficacy. We also discuss the possibility of expanding the biotics family with substances such as bacteriophage, fermented foods, and live dietary microbes.
|
|
Scooped by
?
Today, 1:17 AM
|
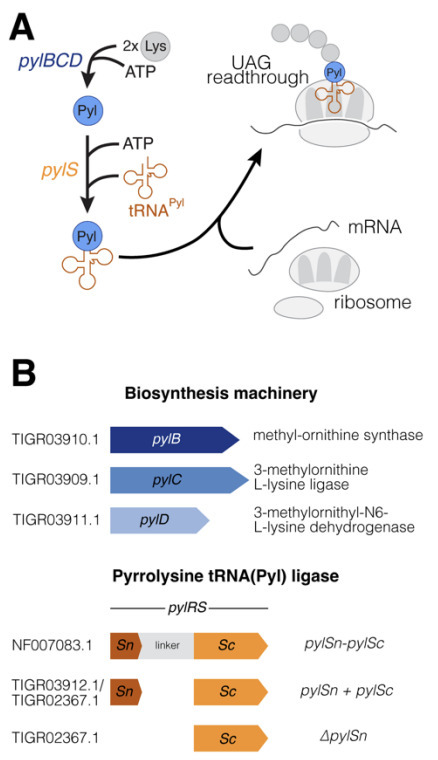
Natural genetic code expansion is a phenomenon wherein an additional amino acid is encoded by a stop codon. These non-standard amino acids are beneficial as they facilitate novel biochemical reactions. However, code expansion leads to ambiguity at the recoded stop codon, which can either be read through or terminated. Pyrrolysine (Pyl) is encoded by the amber codon (TAG/UAG) and is widespread in archaea, where it is required for methylamine-mediated methanogenesis, an environmentally important metabolism. Mechanisms to conditionally suppress the amber stop codon for Pyl installation during protein synthesis have not been identified. Using the model methanogen, Methanosarcina acetivorans, we demonstrate that Pyl-encoding archaea maintain an ambiguous genetic code wherein UAG encodes dual meaning as stop and Pyl. Our data suggest that expression of Pyl biosynthesis and incorporation genes is tuned to the cellular demand for Pyl, which allows these archaea to navigate ambiguous stop decoding in response to environmental cues.
|
|
Scooped by
?
Today, 12:29 AM
|
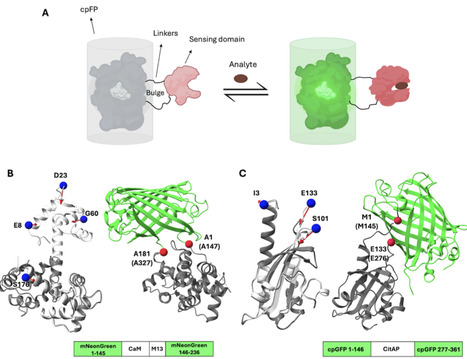
Proteins exhibit remarkable conformational flexibility, enabling precise functional regulation through allostery. A key application of allostery is in the design of protein-based sensors, which detect environmental changes—such as ligand binding or post-translational modifications—and convert these cues into measurable signals (e.g., fluorescence). Here, we investigate a series of ligand-binding proteins that serve as sensing domains in direct-response fluorescent biosensors, wherein ligand binding enhances fluorescence output. We employ a multiple force application approach which we term Multiply Perturbed Response (MPR) to identify “hot spot” residues that drive the conformational transition from an apo (inactive/OFF) to a holo (active/ON) state. We first present two efficient computational approaches to determine residues and forces that maximize the overlap of the observed conformational change. We then determine the overlap maximizer residues for up to five force insertion locations, and we compare them with actual insertion sites used in existing biosensors. Our analysis shows that the allosteric residues identified by MPR coincide with the fluorescent-protein insertion sites that were mapped experimentally through extensive trial-and-error. This work enhances the utility of linear response theory-based methods in uncovering multiple functionally significant regions that trigger a known conformational change. While the validity of the harmonic approximation in anharmonic conformational transitions needs additional validation, MPR gives a good starting point to explore allosteric sites. The approach might prove useful not only in the design of biosensors, but may also find applications in offering physics-based collective variables in mapping conformational transition pathways of proteins.
|
|
Scooped by
?
June 11, 11:50 PM
|
Microbial communities, complex ecological networks crucial for human and planetary health, remain poorly understood in terms of the quantitative principles governing their composition, assembly, and function. Dynamic modeling using ordinary differential equations (ODEs) is a powerful framework for understanding and predicting microbiome behaviors. However, developing reliable ODE models is severely hampered by their nonlinear nature and the presence of significant challenges, particularly critical issues related to identifiability. Here, we address the identification problem in dynamic microbial community models by proposing an integrated methodology to tackle key challenges. Focusing on nonlinear ODE-based models, we examine four critical pitfalls: identifiability issues (structural and practical), unstable dynamics (potentially leading to numerical blow-up), underfitting (convergence to suboptimal solutions), and overfitting (fitting noise rather than signal). These pitfalls yield unreliable parameter estimates, unrealistic model behavior, and poor generalization. Our study presents a comprehensive workflow incorporating structural and practical identifiability analysis, robust global optimization for calibration, stability checks, and rigorous predictive power assessment. The methodology’s effectiveness and versatility in mitigating these pitfalls are demonstrated through case studies of increasing complexity, paving the way for more reliable and mechanistically insightful models of microbial communities. https://doi.org/10.5281/zenodo.15309438
|
|
Scooped by
?
June 11, 11:13 PM
|
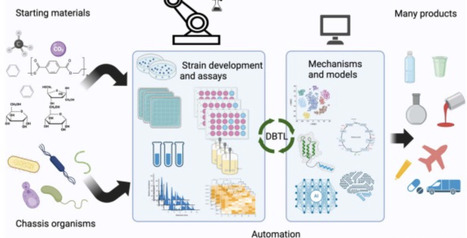
Microbial production of target molecules has advanced significantly in recent years driven by innovations in enzyme engineering, DNA synthesis, and genomic editing. However, to access the massive potential of microbial production, a vast parametric space remains to be investigated to optimize these biobased processes for a robust bioeconomy. Here, we review the current state of the art, some key challenges and possible solutions. We see a critical role of automation, high-throughput technologies, self-driving and cloud labs, and data management to enable Artificial Intelligence/Machine Learning and mechanistic models to overcome the design space challenges and accelerate the development of novel bio-based solutions. Accurate models will expedite the development and scale-up of engineered microbes for a range of final products from many starting materials.
|
|
Scooped by
?
June 11, 10:58 PM
|
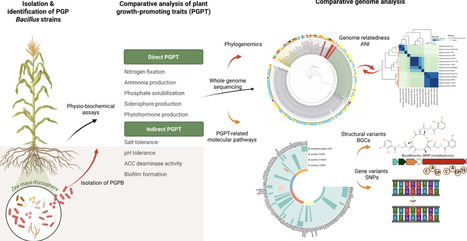
Bacillus species are among the most promising plant growth-promoting bacteria (PGPR) due to their adaptability to various environmental niches and extensive biosynthetic capabilities. Despite the available data on the PGP-traits of Bacillus, the genetic basis underlying their beneficial effects remains largely unexplored. In this study, a comparative genomic analysis of three B. pumilus and one B. pseudomycoides strains, isolated from the maize rhizosphere, is presented to elucidate the molecular mechanisms behind their PGP-traits. All strains exhibited multiple PGP-traits, including phosphate solubilization, phytohormone and siderophore production, growth in nitrogen-free medium, stress tolerance, and biofilm formation. Phylogenomic analysis revealed that plant-associated strains have higher genetic similarity, emphasizing niche-specific evolution. Genome analyses revealed strain-and species-specific adaptations, particularly in relation to nutrient acquisition and abiotic stress response mechanisms. B. pumilus strains encoded alternative sigma factors (SigB, SigM, SigW) enabling enhanced salt tolerance, whereas B. pseudomycoides lacked this system and relied on conventional osmoprotective strategies. The strains utilized different tryptophan-dependent (IAN, IAM or IPyA) pathways for auxin biosynthesis and differed in phosphate solubilization ability, which can be attributed to upstream and missense variants in genes affecting acid metabolism (gltA, acnA, acnB, citM, and citS) and phosphatase (phoA) activity. Iron uptake via bacillibactin-siderophores was exclusive to B. pumilus. The inability of the B. pseudomycoides strain to acquire iron was associated with structural variants (absence of bsaA gene) within the bacillibactin biosynthetic gene cluster. This work provides new insights into the molecular basis of PGP traits in Bacillus and supports the development of Bacillus-based bioinoculants for sustainable agriculture.
|
|
Scooped by
?
June 11, 7:29 PM
|
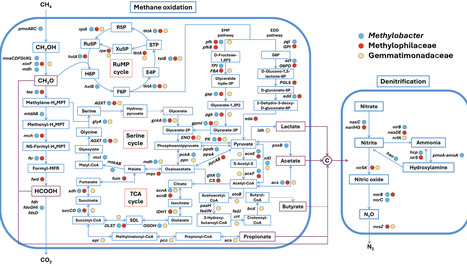
The impact of anaerobic oxidation of methane (AOM) coupled with denitrification on the emission of the denitrification intermediate N2O remains poorly understood. Here, we investigated the influence of AOM coupled with nitrate and nitrite reduction on soil N2O emissions and the associated microbial interactions. We show that AOM coupled with denitrification markedly reduces soil N2O emissions, with the type I methanotroph Methylobacter species collaborating with the Methylophilaceae and Gemmatimonadaceae families to perform key roles. The suppression of N2O emissions by AOM primarily stems from its role in supplying electrons and carbon sources, fueling complete denitrification by associated bacteria. In addition, we uncovered distinct microbial interaction strategies for AOM-coupled nitrate and nitrite reduction. While nitrite reduction necessitates both bacterial cooperation and extracellular electron transfer during its initial stages, nitrate reduction predominantly depends on methanotrophic bacteria alone at the outset. These findings advance our understanding of carbon-nitrogen cycle coupling and underscore AOM’s potential to simultaneously mitigate both CH4 and N2O emissions.
|
|
Scooped by
?
June 11, 6:50 PM
|
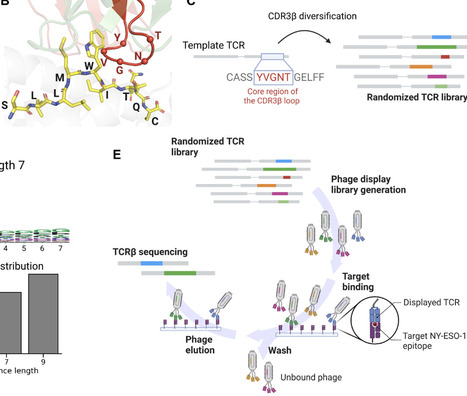
T cells targeting epitopes in infectious diseases or cancer play a central role in spontaneous and therapy-induced immune responses. Epitope recognition is mediated by the binding of the T cell receptor (TCR), and TCRs recognizing clinically relevant epitopes are promising for T cell–based therapies. Starting from a TCR targeting the cancer-testis antigen NY-ESO-1157–165 epitope, we built large phage display libraries of TCRs with randomized complementary determining region 3 of the β chain. The TCR libraries were panned against NY-ESO-1, which enabled us to collect thousands of epitope-specific TCR sequences. Leveraging these data, we trained a machine learning TCR-epitope interaction predictor and identified several epitope-specific TCRs from TCR repertoires. Cellular assays revealed that the predicted TCRs displayed activity toward NY-ESO-1 and no detectable cross-reactivity. Our work demonstrates how display technologies combined with TCR-epitope interaction predictors can effectively leverage large TCR repertoires for TCR discovery.
|
|
Scooped by
?
June 11, 6:20 PM
|
Cas12j-8 is a compact Cas nuclease discovered from the metagenome of giant bacteriophages, consisting of only 717 amino acids and recognizing the ‘5-TTN-3′ protospacer adjacent motif (PAM) sequence. However, its low gene editing efficiency in mammalian cells limits its application in therapeutic gene editing. To address this limitation, structure-guided mutagenesis is employed to replace key negatively charged residues with arginine, strengthening DNA binding. The resulting quintuple mutant, engineered Cas12j-8 (enCas12j-8), demonstrates robust on-target editing efficiency comparable to LbCas12a while maintaining low off-target effects. Cytosine base editors (CBEs) and adenine base editors (ABEs) are developed using enCas12j-8, achieving up to 29.54-fold C-to-T and 36.57-fold A-to-G conversion efficiency compared with the wild-type at the dominated sites, respectively. Notably, enCas12j-8 enables multiplexed editing of three genomic loci simultaneously via a single crRNA array, achieving efficiencies comparable to single-guide approaches. Additionally, enCas12j-8-ABE facilitates the disruption of splice acceptor sites, effectively inducing exon skipping in the SOD1 gene. This strategy holds potential significance for therapeutic genome modulation. These findings establish enCas12j-8 as a versatile, high-precision tool for genome engineering, combining efficient delivery, multiplexing capability, and compatibility with diverse editing modalities.
|















sattely es, Biochemical breakthrough paves way to reliable supply of anticancer drug https://www.nature.com/articles/d41586-025-01458-5