 Your new post is loading...

|
Scooped by
?
Today, 3:57 PM
|
Widespread application of bacterial-based cancer therapy is limited because of the need to increase therapeutic bacteria specificity to the tumor to improve treatment safety and efficacy. Here, we harness the altered tumor metabolism and specifically elevated kynurenine accumulation to target engineered bacteria to the cancer site. We cloned and leveraged kynurenine-responsive transcriptional regulator (KynR) with its cognate promoter in Escherichia coli. Optimizing KynR expression coupled with overexpressing kynurenine transporter and amplifying the response through plasmid copy number–based signal amplification enabled the response to kynurenine at the low micromolar levels. Knocking out genes essential for cell wall synthesis and supplying these genes via kynurenine-controlled circuits allowed tuning Salmonella enterica growth in response to kynurenine. Our kynurenine-controlled S. enterica (hereafter named AD95+) showed superior tumor specificity in breast and ovarian cancer murine models compared to S. enterica VNP20009, one of the best characterized tumor-specific strains. Last, AD95+ showed anticancer properties compared to vehicle controls, demonstrating the potential as an anticancer therapeutic.
|
|
Scooped by
?
Today, 11:19 AM
|
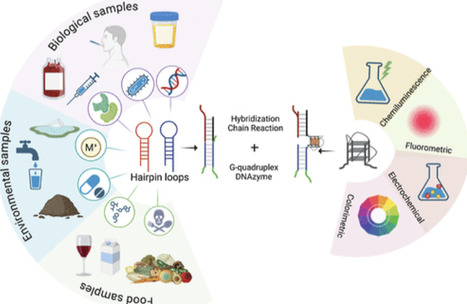
The hybridization chain reaction (HCR) is an amplification method recognized for detecting analytes present in trace quantities owing to its high specificity, sensitivity, and straightforward approach. Simultaneously, G-quadruplex DNAzymes augment HCR-based biosensors, serving as transducers due to their elevated catalytic activity and nonenzymatic methodology. The current review aims to provide readers with a critical overview of significant aspects of biosensors that utilize HCR for analyte detection amplification and G-quadruplex as transducers, focusing on the latest activities of global researchers. A bibliometric analysis was conducted to identify key research topics in HCR and G-quadruplex-based sensors. Further, the review aims to elucidate the advantages and disadvantages of each strategy employed for detection using this sort of sensor. Specific sections have been included to critically evaluate the potential of these biosensors across many scientific domains. In conclusion, the limitations of the existing sensor that restrict its applicability in real-time scenarios have been identified for subsequent enhancement. Plausible strategies that could enhance these sensors and emerging scientific fields that could benefit from HCR and G-quadruplex DNAzyme-based sensors have been elucidated.
|
|
Scooped by
?
Today, 10:41 AM
|
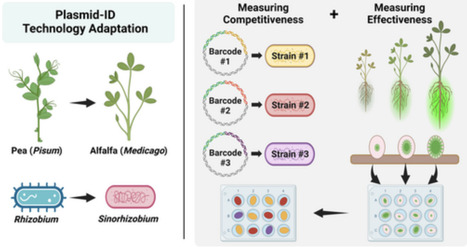
Maximizing the nitrogen fixation occurring in rhizobia-legume associations represents an opportunity to sustainably reduce nitrogen fertilizer inputs in agriculture. High-throughput measurement of symbiotic traits has the potential to accelerate the identification of elite rhizobium/legume associations and enable novel research approaches. Plasmid-ID technology, recently deployed in Rhizobium leguminosarum, facilitates the concurrent assessment of rhizobium nitrogen-fixing effectiveness and competitiveness for root nodulation. This study adapts Plasmid-ID technology to function in Sinorhizobium species that are central models for studying rhizobium-legume associations and form economically important symbioses with alfalfa. New Sino-Plasmid-IDs were developed and tested for stability and their ability to measure competitiveness for root nodulation and nitrogen-fixing effectiveness. Rhizobial competitiveness is measured by identifying strain-specific nucleotide barcodes using next-generation sequencing, whereas effectiveness is measured by GFP fluorescence driven by the synthetic nifH promoter. Sino-Plasmid-IDs allow researchers to efficiently study competitiveness and effectiveness in a multitude of Sinorhizobium strains simultaneously.
|
|
Scooped by
?
Today, 10:37 AM
|
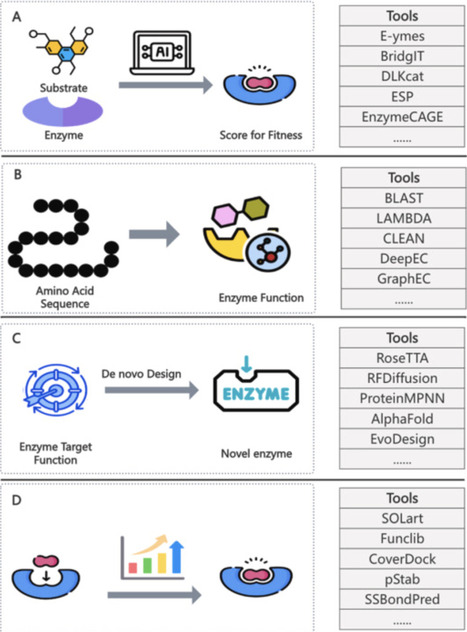
One of the main goals in synthetic biology is to produce value-added compounds from available precursors using enzymatic approaches. The construction of biosynthetic pathways for synthesizing target molecules plays a crucial role in this process. However, it is challenging and time-consuming for researchers to design efficient pathways manually. In recent decades, pathway design has advanced through data- and algorithm-driven approaches. In this article, we review key computational tools involved in biosynthetic pathway design, covering: 1) Biological Big-Data including compounds, reactions/pathways and enzymes. 2) Retrosynthesis methods leveraging multi-dimensional biosynthesis data to predict potential pathways for target compounds synthesis. 3) Enzyme engineering relying on data mining to identify/de novo design enzymes with desired functions. Integrating these three key components can significantly enhance the efficiency and accuracy of biosynthetic pathway design in synthetic biology.
|
|
Scooped by
?
Today, 10:18 AM
|
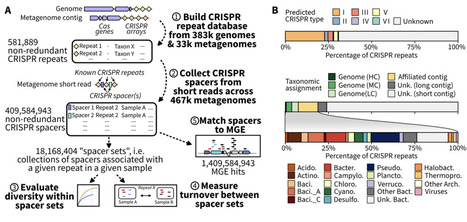
Interactions between microbes and their mobile genetic elements (MGEs), including viruses and plasmids, are critical drivers of microbiome structures and processes. CRISPR-Cas systems are known to be important regulators of these host-MGE interactions, but a global understanding of CRISPR-Cas diversity, activity, and roles across Earth's biomes is still lacking. Here, we use an optimized computational approach to search short-read data and collect ~800 million CRISPR spacers across ~450,000 public metagenomes. Comparing spacers across samples and taxa revealed a high population diversity for CRISPR loci overall, with typically only a small subset of spacers detected as prevalent and conserved within a microbial population. From this extensive CRISPR spacer dataset, we identified 1.18 billion hits between 41 million spacers and 2.5 million viruses and plasmids. Prevalent and conserved spacers were over-represented in these MGE-matching spacers, and CRISPR spacers frequently matched multiple MGEs, consistent with a positive selection pressure associated with MGE targeting. Focusing on the role of CRISPR as antiphage defense, we observed surprising cases of viruses targeted by microbes not expected to be viable hosts. These were more frequent for viruses encoding a diversification mechanism (DGR) for their host attachment proteins, and associated with a reduced rate of escape mutations. This suggests that broad spacer targeting may derive from the recurring entry of a virus genome into a non-host microbial cell, leading to some viruses being targeted by taxonomically diverse microbes well outside of their actual host range. Taken together, this petabase-scale exploration of CRISPR arrays in nature outlines the extensive diversity of CRISPR array loci across microbiomes. It highlights several key genomic and ecological parameters driving the activity of CRISPR arrays that are likely influencing strain-level diversification and selection processes within microbial populations.
|
|
Scooped by
?
Today, 9:56 AM
|
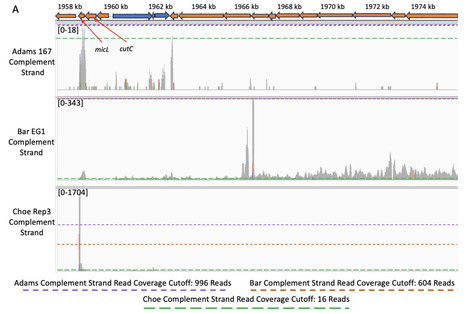
3-prime end sequencing (3′-seq) is a high-throughput sequencing technique that is used to specifically quantify the changes in 3′-end formation of transcripts in bacterial cells, which is increasingly being utilized to address fundamental questions regarding transcription termination and pausing across a range of different bacterial species. However, the growing number of 3′-seq studies is accompanied by an increase in study-specific 3′-seq data analysis approaches. Thus, differences in a number of factors including: experimental design, data collection approaches, analysis methodologies, and interpretation decisions, make it challenging to confidently compare results derived from different studies, even those that were performed on the same organism. To assess the potential severity of these discrepancies, we used PIPETS, a statistically robust and genome-annotation agnostic 3′-seq analysis package, to study Escherichia coli 3′-seq data sets from three different groups collected under similar conditions. By using a consistent analysis and results interpretation approach, we identified large disparities in the characteristics of the raw 3′-seq data between each of the studies, despite all three studies using the same strain and very similar reported experimental conditions. Additionally, we found strand-specific inconsistencies, with some data sets having reference strand 3′-seq read coverage distributions that differed greatly from the complement strand within the same replicate. Finally, when the 3′-seq distribution profiles of the three E. coli studies are compared to studies from four additional bacteria, we identified 3′-seq results clustering patterns that are not explained by phylogenetic similarity between organisms. With the large differences seen between data sets from the same organism as well as the inconsistencies seen between replicates from the same data sets, we urge the field to reconsider the assumptions around 3′-seq data homogeneity and move towards consistent analysis approaches, and cautious interpretation of the data.
|
|
Scooped by
?
Today, 1:42 AM
|
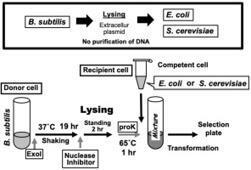
Purified DNA plasmids traditionally used for microbial transformation have been supplanted by extracellular plasmids released via host bacterial lysis, offering an alternative approach for DNA-plasmid delivery. Specifically, shuttle vector plasmids liberated from host Bacillus subtilis were directly employed for the transformation of chemically competent cells Escherichia coli, eliminating the need for biochemical purification. This unconventional DNA delivery technique, referred to as ’Cell Lysis Technology to provide Transformable Extra-cellular DNA; CELyTED’, has been successfully adapted for the transformation of microorganism Saccharomyces cerevisiae as well. The protocol includes optimized conditions for efficient cell lysis of the donor host cells. Notably, ’ CELyTED ’ enables the introduction of large-sized DNA plasmids exceeding 50 kb into target microorganisms mitigating the potential adverse effects of physical shearing during the purification process. This simplicity in the delivery protocol makes it versatile for both prokaryotic and eukaryotic microorganisms, establishing a fundamental platform in the synthetic genome field. Our study demonstrates the feasibility of introducing large DNA plasmids into cells E. coli and S. cerevisiae using the lysate of donor host cells, showcasing the potential of ‘CELyTED ’ as a streamlined approach in genetic transformation methodologies.
|
|
Scooped by
?
Today, 1:01 AM
|
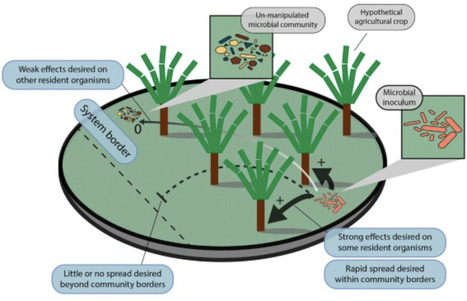
Microbial inoculants are increasingly used for beneficial purposes in agriculture, bioremediation, and medicine, but they can carry risks of generating invasive microbes. Here, we present a roadmap for guarding against these invasions, proposing developing (i) coherent mechanistic understandings of how microbial inoculants can effect invasions, (ii) predictive models forecasting microbial invasion risks, and (iii) effective management strategies. To guide mechanistic understandings, we distill 17 guiding hypotheses. For predictive modeling, we highlight data collection needs and qualitative approaches. For management strategies, we stress the importance of accurately weighing the risks against benefits. The unified approach presented here provides a route toward an effective research and management infrastructure for microbial inoculants in order to avoid potentially catastrophic microbial invasions.
|
|
Scooped by
?
Today, 12:09 AM
|
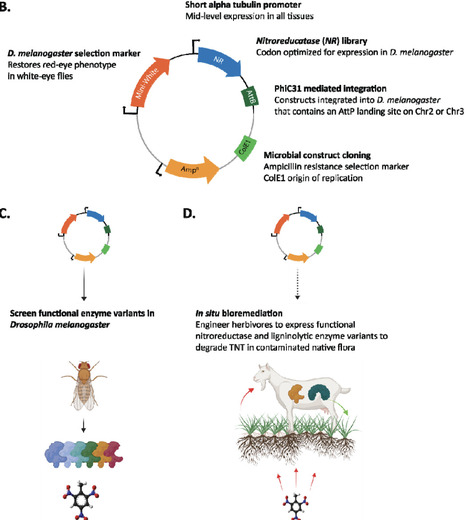
TNT (2,4,6-trinitrotoluene) from unexploded ordinances is a common environmental pollutant near munitions factories, military training sites, and areas of armed conflict. As physical and chemical approaches for TNT remediation are costly and difficult to scale, in situ bioremediation is an attractive alternative. TNT is a phytoaccumulative pollutant. Herbivores engineered to detoxify TNT are a potentially cost-effective platform to bioremediate large areas of contaminated sites while grazing or browsing. As a first step towards engineering herbivores with metabolic capabilities for TNT degradation, we engineered the animal genetic model, Drosophila melanogaster, to screen a library of microbial nitroreductases that can catalyse the initial degradation steps required for TNT degradation. We find strong, cofactor-dependent activity in fly lysates engineered to express NsfA from Escherichia coli, and NsfI from Enterobacter cloacae. bioremediation
|
|
Scooped by
?
June 12, 11:41 PM
|
Cas1 and Cas2 are the hallmark proteins of prokaryotic adaptive immunity. However, these two proteins are often fused to other proteins and the functional association of these fusions often remain poorly understood. Here we purify Cas1 and the Cas2/3 fusion protein from Pseudomonas aeruginosa. We determine multiple structures of the Cas1-2/3 complex at distinct stages of CRISPR adaptation. Collectively, these structures reveal a prominent, positively charged channel on one face of the integration complex that captures short fragments of foreign DNA. Foreign DNA binding triggers conformational changes in Cas2/3 that expose new DNA binding surfaces necessary for homing the DNA-bound integrase to specific CRISPR loci. The length of the foreign DNA substrate determines if Cas1-2/3 docks completely onto the CRISPR repeat to successfully catalyze two sequential transesterification reactions required for integration. Taken together, these structures clarify how the Cas1-2/3 proteins orchestrate foreign DNA capture, site-specific delivery, and integration of new DNA into the bacterial genome.
|
|
Scooped by
?
June 12, 11:03 PM
|
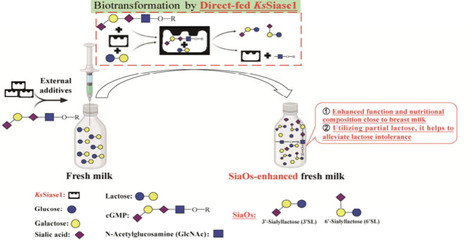
Sialyloligosaccharides, critical components of human milk oligosaccharides, are pivotal for infant immunity and development. Recent advances have identified sialidases with trans-sialylation activity as promising biocatalysts for cost-effective sialyloligosaccharide synthesis using economical substrates. Herein, we report a novel sialidase (KsSiase1) from Kribbella sp. ALI-6-A with enhanced catalytic properties and broad substrate specificity, showing preferential hydrolysis: α2,3 > α2,6 > α2,8. The enzyme demonstrated robust trans-sialidase activity, achieving 3.40 g/L sialyloligosaccharide production using 0.5 U/mL KsSiase1, 6% lactose, and 8% cGMP. Implementing a direct-fed enzymatic strategy in raw milk with 1 U/mL KsSiase1 and 6% cGMP, we achieved in situ sialyloligosaccharide enrichment (2.49 g/L) while concomitantly reducing lactose. This biocatalytic system establishes a sustainable, scalable platform for functional food production, merging lactose valorization with sialyloligosaccharides biosynthesis through enzyme–substrate adaptability, addressing both industrial demands and nutritional needs in infant formula development.
|
|
Scooped by
?
June 12, 10:53 PM
|
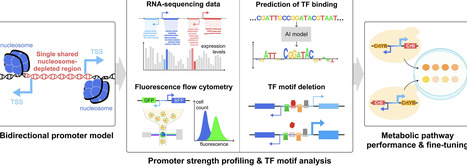
Bidirectional promoters (BDPs) hold great promise for applications in synthetic biology by enabling co-expression of multiple genes with minimized promoter size. However, the lack of well-characterized BDPs along with an incomplete understanding of their regulatory mechanisms limits broader applications. Here, we conducted genome-wide screening and characterization of 749 BDP candidates containing a single shared nucleosome-depleted region in yeast Saccharomyces cerevisiae. A pronounced asymmetry in BDP strength was observed using both transcriptomic and fluorescence reporter analyses. We demonstrated that these unbalanced BDP strengths could be utilized for fine-tuning metabolic flux in yeast, achieving yields comparable to or exceeding those of commonly used constitutive or inducible promoters for terpenoid production under the examined conditions. Using in silico mutagenesis guided by the DREAM-CNN yeast cis-regulatory AI prediction model, we identified conserved activator-binding hotspots within the central region of 63.8% of identified BDP candidates. Disruption of these hotspots in six selected BDPs significantly reduced promoter strength in both orientations, suggesting that these AI-predicted motifs are indeed critical for the functionality of BDPs. Overall, this study provides a comprehensive framework for BDP identification and engineering, leveraging AI-guided models to advance rational synthetic promoter design, thus paving the way for precise genetic control in synthetic biology.
|
|
Scooped by
?
June 12, 4:37 PM
|
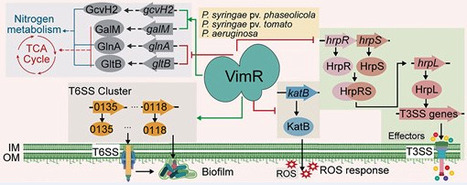
Pseudomonas syringae, a highly destructive plant bacterial pathogen causing severe disease and significant yield losses in agriculture globally, has complex regulatory systems involving many transcriptional factors (TFs). Although the LysR-type transcriptional regulator (LTTR) protein family is a well-known group of TFs involved in diverse physiological functions, the roles of LTTRs in P. syringae remain largely unknown. In this study, we characterized a LysR-type TF, PSPPH4638, and designated it as the virulence and metabolism regulator VimR. Genome-wide identification of VimR using chromatin immunoprecipitation sequencing revealed 1032 binding sites in the genome, of which 85% were in intergenic regions. Transcriptomic analysis showed altered expression of 454 and 82 genes in response to ΔvimR in King’s B medium (KB) and minimal medium (MM), respectively. Conjoint analysis showed that 99 genes were directly affected by VimR in KB. VimR was identified as a repressor of the type III secretion system, oxidative stress response, and key metabolic pathways such as the tricarboxylic acid cycle. In addition, we found that VimR was positively involved in the type VI secretion system and alanine, aspartate, and glutamate metabolism. Further verification showed that VimR was widely present in Pseudomonas, displaying similar binding capacity in different strains of P. syringae, and similar regulatory functions in Pseudomonas aeruginosa. Taken together, our findings identified a conserved master TF that regulates type III secretion system, type VI secretion system, and multiple metabolic pathways in Pseudomonas.
|
|
|
Scooped by
?
Today, 3:25 PM
|
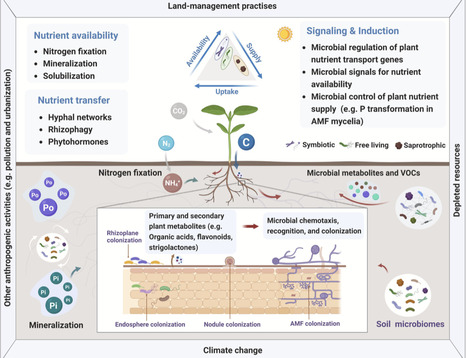
Root nodule symbiosis allows for plant acquisition of reactive nitrogen through fixation of atmospheric molecular dinitrogen by nitrogen-fixing bacteria. Nodulation is a complex trait, with diverse modes of bacterial infection and nodule morphologies across species, reflecting evolutionary adaptation. Understanding ancient forms of this trait may carry advantages for its current utilization, since basal states likely reflect the least complexity. In this review we focus on the evolution of nodule development, particularly on events that have led to increased complexity of this symbiosis in later adaptations. We hypothesize that the ancestral form of nodulation comprises of an evolutionary coupling of nutrient-dependent lateral root development with apoplastic intercellular bacterial growth, alongside the acquisition or evolution of an ancestral chitinaceous signaling molecule by the microbial symbiont. Uncovering the evolutionary adaptations underpinning the extant diversity of this trait allows for a better understanding of the simplest ancestral state.
|
|
Scooped by
?
Today, 10:46 AM
|
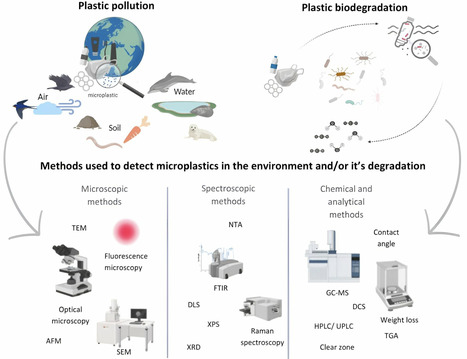
Environmental concerns about the widespread use of non-biodegradable plastic have generated interest in developing quick and effective methods to degrade synthetic polymers. With millions of tons of plastic waste generated annually, biodegradation by microorganisms presents a promising and eco-friendly solution. However, a bottleneck has arisen due to the lack of standardized methods for verification of the biodegradation process. Based on this literature review, he techniques most commonly employed for this purpose currently include measuring mass loss, examining the surface of plastic fragments by scanning electron microscopy (SEM) and atomic force microscopy (AFM), and using analytical methods such as Fourier transform infrared spectroscopy (FTIR), pyrolysis–gas chromatography–mass spectrometry (Pyr-GC/MS) or high-performance liquid chromatography (HPLC). Each of these methods has its advantages and disadvantages. Nevertheless, currently, there is no universal approach to accurately assess the ability of individual microorganisms to degrade plastics. In this review, we summarize the latest advances in techniques for detecting biodegradation of synthetic polymers and future directions in the development of sustainable strategies for mitigating plastic pollution.
|
|
Scooped by
?
Today, 10:39 AM
|
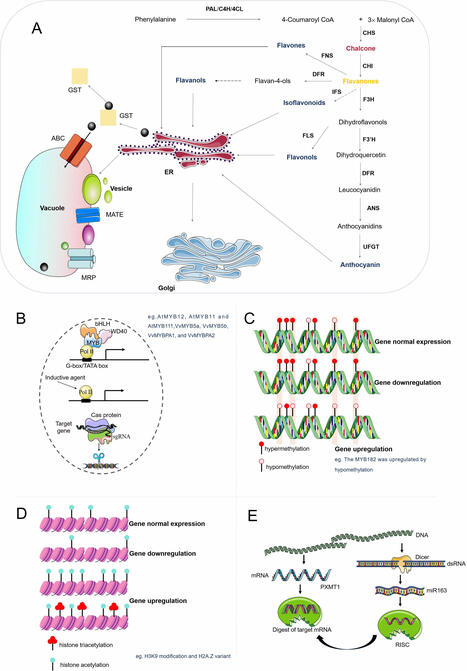
Flavonoids are a diverse class of plant polyphenols with essential roles in development, defense, and environmental adaptation, as well as significant applications in medicine, nutrition, and cosmetics. However, their naturally low abundance in plant tissues poses a major barrier to large-scale utilization. This review provides a comprehensive and forward-looking synthesis of flavonoid biosynthesis, regulation, transport, and yield enhancement strategies. We highlight key advances in understanding transcriptional and epigenetic control of flavonoid pathways, focusing on the roles of MYB, bHLH, and WD40 transcription factors and chromatin modifications. We also examine flavonoid transport mechanisms at cellular and tissue levels, supported by emerging spatial metabolomics data. Distinct from conventional reviews, this review explores how plant cell factories, genome editing, environmental optimization, and artificial intelligence (AI)-driven metabolic engineering can be integrated to boost flavonoid production. By bridging foundational plant science with synthetic biology and digital tools, this review outlines a novel roadmap for sustainable, high-yield flavonoid production with broad relevance to both research and industry.
|
|
Scooped by
?
Today, 10:36 AM
|
Plants have evolved complex regulatory mechanisms to cope with environmental challenges. Recent work reveals how distinct root cell types coordinate their specialized genetic responses to form diffusion barriers and adapt to soil stress. In non-compacted soil, water in the soil is absorbed by the roots and transported upwards through the vasculature to support plant hydration and physiological processes. In compacted soil, water stress is exerted on roots as moisture release is reduced from the smaller soil pores. Abscisic acid (ABA) synthesis is activated in the vasculature via unknown genetic mechanisms involving complex hormonal crosstalk with other plant hormones such as ethylene and auxin; the ABA then moves outwards with water flux to activate lignin and suberin deposition in outer tissues. These newly deposited barriers collectively act to prevent root water loss by reducing radial water efflux, preserving internal water status.
|
|
Scooped by
?
Today, 10:02 AM
|
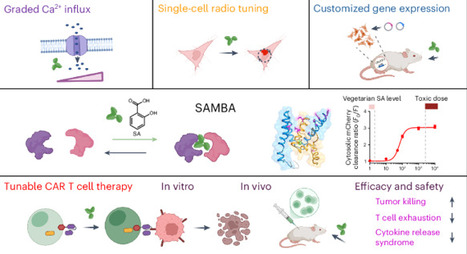
Chemically induced proximity (CIP) has remarkably advanced the development of molecular and cellular therapeutics. To maximize therapeutic potential, there is a pressing need to expand the repertoire of CIP systems of translational values, favoring chemical ligands that are cost-effective, structurally simple, biocompatible, reversible and have minimal side effects. Here, we present a salicylic acid (SA)-mediated binary association system (SAMBA), evolved from a tobacco SA receptor, that enables rapid protein–protein heterodimerization in response to SA or aspirin after hydrolysis. We demonstrate the broad applicability of SAMBA in various biological contexts, including SA-dependent reprogramming of a protein-based reaction–diffusion system, graded gating of calcium channels, inducible initiation of receptor tyrosine kinase-mediated signaling and gene expression, and tunable activation of chimeric antigen receptor T cells. Our work establishes SAMBA as a versatile chemogenetic platform that allows temporal control of biological processes and therapeutic cells both in vitro and in vivo. A highly compact chemically induced proximity system dependent on salicylic acid or aspirin enables control of biological processes and cellular therapeutics using an over-the-counter drug with minimal side effects.
|
|
Scooped by
?
Today, 1:44 AM
|
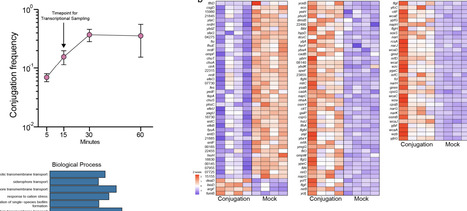
Conjugative plasmids drive bacterial evolution and niche adaptation. Their carriage often corresponds with changes to the host transcriptome, but it is unknown during which stage of the conjugation process these transcriptional changes are initiated, and how gene transfer is modulated as they occur. This is because they are normally studied after their full acquisition by conjugation has already occurred, not during the process. Here, active RP4 conjugation revealed that the transcriptional response to plasmid acquisition is almost immediate and is host and surface factor dependent. Responses included activation of non-SOS stress pathways, motility, exopolysaccharide production, anaerobic respiration, and metabolic adaptation. These responses were not a barrier to conjugation, indicating that their significance relates to host homeostasis rather than gene transfer. The recipient could prevent these responses by blocking conjugation through capsule expression. RP4 expression was also inhibited by recipient capsule, since capsule physically blocked donor-recipient contact. Unexpectedly, within a population, RP4 was found to exploit single cell variation in recipient capsule thickness and to successfully conjugate with recipient bacteria with reduced capsule. These results show a novel interplay between plasmid and surface architecture across diverse species that controls the conjugation transcriptional landscape, with important ramifications for plasmid dissemination and evolution.
|
|
Scooped by
?
Today, 1:09 AM
|
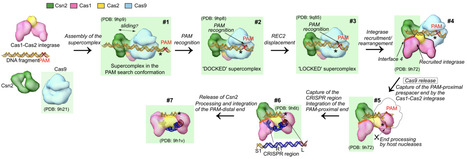
During CRISPR-Cas adaptation, prokaryotic cells become immunized by the insertion of foreign DNA fragments, termed spacers, into the host genome to serve as templates for RNA-guided immunity. Spacer acquisition relies on the Cas1-Cas2 integrase and accessory proteins like Cas4, which select DNA sequences flanked by the protospacer adjacent motif (PAM) and insert them into the CRISPR array. It has been shown that in type II-A systems selection of PAM-proximal prespacers is mediated by the effector nuclease Cas9, which forms a 'supercomplex' with the Cas1-Cas2 integrase and the Csn2 protein. However, the supercomplex structure and the role of the ring-like Csn2 protein remain unknown. Here, we present cryo-electron microscopy structures of the type II-A prespacer selection supercomplex in the DNA-scanning and two different PAM-bound configurations. Our study uncovers the mechanism of Cas9-mediated prespacer selection in type II-A CRISPR-Cas systems, and reveals the role of the accessory protein Csn2, which serves as a platform for the assembly of Cas9 and Cas1-Cas2 integrase on prespacer DNA, reminiscent of the sliding clamp in DNA replication. Repurposing of Cas9 by the CRISPR adaptation machinery for prespacer selection characterized here demonstrates Cas9 plasticity and expands our knowledge of the Cas9 biology.
|
|
Scooped by
?
Today, 12:54 AM
|
Regulatory elements are essential components of plant genomes that have shaped the domestication and improvement of modern crops. However, their identity, function and diversity remain poorly characterized, limiting our ability to harness their full power for agricultural advances using induced or natural variation. Here we mapped transcription factor (TF) binding for 200 TFs from 30 families in two distinct maize inbred lines historically used in maize breeding. TF binding comparison revealed widespread differences between inbreds, driven largely by structural variation, that correlated with gene expression changes and explained complex quantitative trait loci such as Vgt1, an important determinant of flowering time, and DICE, an herbivore resistance enhancer. CRISPR–Cas9 editing of TF binding regions validated the function and structure of regulatory regions at various loci controlling plant architecture and biotic resistance. Our maize TF binding catalogue identifies functional regulatory regions and enables collective and comparative analysis, highlighting its value for agricultural improvement. Genome-wide comparative analysis of DNA binding for 200 transcription factors in two maize inbred lines reveals prevalent genotype-specific variation that is associated with gene expression differences and agronomic traits.
|
|
Scooped by
?
June 12, 11:56 PM
|
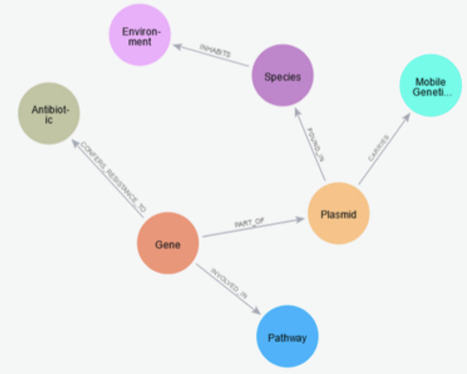
Horizontal gene transfer (HGT) is a key mechanism driving the rapid dissemination of antimicrobial resistance (AMR) among bacterial populations, posing a severe threat to global public health. Traditional detection methods for HGT, such as phylogenetic incongruence and compositional analysis, are often limited in their scope and accuracy. To address these challenges, we propose a comprehensive knowledge graph (KG)-based framework that integrates genomic, environmental, biochemical, and ontological datasets to detect and analyze HGT events contributing to AMR. Our KG captures relationships among genes, plasmids, species, mobile genetic elements, antibiotics, and ecological factors using curated sources like NCBI, CARD, ICEberg, MGnify, and KEGG. We enrich nodes and edges with biological (e.g., GC content, codon bias) and chemical (e.g., binding affinities, environmental exposure) signatures and apply ontology-based semantic enrichment using ARO and GO. A Graph Neural Network (GNN) trained on the KG achieves high accuracy (92%) and F1-score (0.90) in predicting HGT events, outperforming conventional methods. Community detection identifies AMR dissemination hubs, while environmental analyses reveal strong correlations between antibiotic concentrations and resistance gene prevalence. Our framework demonstrates strong biological and chemical validation and provides a scalable, interpretable, and data-rich tool for AMR surveillance, with implications for clinical, agricultural, and environmental microbiology.
|
|
Scooped by
?
June 12, 11:21 PM
|
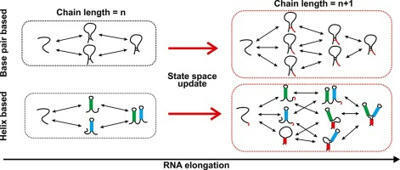
An RNA folds as it is transcribed. RNA folding during transcription differs fundamentally from thermodynamic folding. While thermodynamic folding reaches an equilibrium ensemble of structures, cotranscriptional folding is a kinetic process where the RNA structure evolves as the chain elongates during transcription. This dynamic folding pathway causes cotranscriptional structures often to deviate from thermodynamic predictions, as the system rarely reaches equilibrium. Since these cotranscriptional effects can persist in the mature RNA's structure, understanding this kinetic process is crucial. While experimental studies of cotranscriptional folding have been successful, they remain resource-intensive. Computational modeling has emerged as an increasingly practical and powerful approach for investigating these dynamics. This short review examines current computational methods and tools for simulating cotranscriptional folding, with the goal of advancing our understanding of RNA folding mechanisms.
|
|
Scooped by
?
June 12, 11:00 PM
|
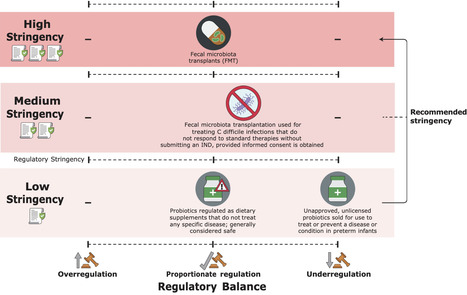
The three main types of live bacterial therapies – probiotics, fecal/microbiome transplants, and engineered bacterial therapies – hold immense potential to revolutionize medicine. While offering targeted and personalized treatments for various diseases, these therapies also carry risks such as adverse immune reactions, antibiotic resistance, and the potential for unintended consequences. Therefore, developing and deploying these therapies necessitates a robust regulatory framework to protect public health while fostering innovation. In this paper, we propose a novel conceptual tool – the Ladder of Regulatory Stringency and Balance—which can assist in the design of robust regulatory regimes which encompass medicine practices based not only on definitive Randomized Controlled Trials (RCTs), but also on meta-analyses, observational studies, and clinicians experience. Regulatory stringency refers to the strictness of regulations, while regulatory balance concerns the degree of alignment between the regulatory framework governing a technology and the actual risks posed by specific products within that technology. Focusing on the US regulatory environment, we subsequently position the three types of live bacterial therapies on the Ladder. The insight gained from this exercise demonstrates that probiotics are generally positioned at the bottom of the Ladder, corresponding to low-stringency regulation, with a proportionate regulatory balance. However, probiotics intended for high-risk populations are currently subject to low-stringency regulations, resulting in under-regulation. Our analysis also supports the conclusion that fecal microbiota transplants (FMT) for recurrent Clostridium difficile infection should be positioned close to but below the threshold for under regulation by the U.S. Food and Drug Administration (FDA), and we recommend improved donor screening procedures, preservation and processing, storage, and distribution. Our framework can serve as a scale to assess regulatory gaps for live bacterial therapies and to identify potential solutions where such gaps exist.
|
|
Scooped by
?
June 12, 10:43 PM
|
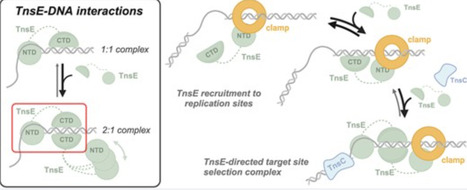
Tn7 transposable elements are known for their sophisticated target-site selection mechanisms. For the prototypical Tn7 element, dedicated transposon-encoded proteins direct insertions to either a conserved site in the chromosome or replicating DNA structures in conjugal plasmids, ensuring the vertical and horizontal spread of the element. While the pathway targeting the attTn7 site in the bacterial chromosome has been extensively studied, the pathway targeting DNA replication structures remains poorly understood. We have used an integrative structural biology approach to elucidate how the Tn7-encoded protein TnsE recognizes replication sites. Using native mass spectrometry, we found that TnsE forms 1:1 and 2:1 (TnsE:DNA) complexes on 3′-recessed DNA, with gain-of-function TnsE variants favoring the formation of 2:1 complexes. Structural characterization confirms that two TnsE molecules bind to DNA with the C-terminal domain of the protein recognizing duplex DNA, leaving the N-terminal domain to impose DNA substrate specificity and recruit the core transposition machinery. Collectively, our work is consistent with a model where TnsE-mediated target-site selection relies on the formation of an asymmetric TnsE:DNA complex to recruit the Tn7 transposase to DNA replication structures.
|















singh bk