Your new post is loading...

|
Scooped by
?
December 13, 7:30 PM
|
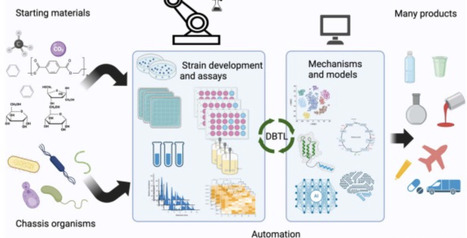
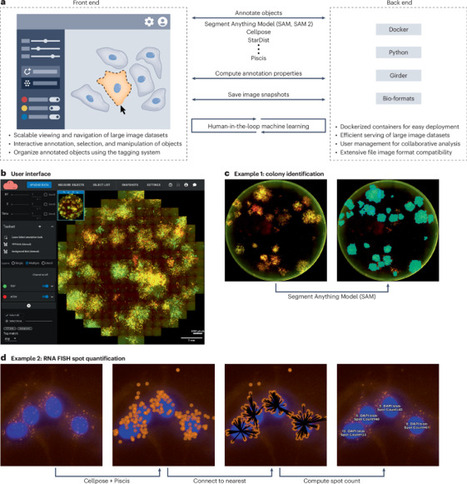
Imaging has been critical to biological discoveries for centuries. Throughout, one of the primary challenges has been the quantitative analysis of these images. The solutions to date have largely involved custom software in combination with software packages such as ImageJ1, napari2, CellProfiler3, MATLAB, and others. However, there remains a need for users to interact with their data even if they lack the ability to code. New machine-learning algorithms have the potential to scale our ability to accurately quantify imaging data, but the technical expertise required to deploy these tools puts them out of reach for many users. Here, we introduce NimbusImage, a software package that addresses these challenges. NimbusImage brings advancements in image analysis to users who may otherwise find such tools difficult to use, all in an easy-to-use web-based platform. Key features include cloud-based deployment, an intuitive interface that enables direct interaction with data, an extensible API (application programming interface), and plug-ins that combine conventional analytical methods with newer deep-learning techniques.
|
|
Scooped by
?
December 13, 7:26 PM
|
The rhizosphere microbiome directly influences plant health and acclimation to extreme environments, yet plant-microbe interactions in the rhizosphere have proven complex and difficult to study. We present RhizoGrid, a new methodological framework that integrates a 3D-printed pot structure with spatial measurements of root structure, metabollite and taxonomy to detect links between metabolites and microbes along a soil-grown root system. The RhizoGrid identifies microhabitats hidden belowground. Using the food, forage, and bioenergy crop sorghum, we showcase how the RhizoGrid opens new frontiers for exploring the heterogeneity and complexity of interactions between root exudates and microbes in the rhizosphere.
|
|
Scooped by
?
December 13, 7:18 PM
|
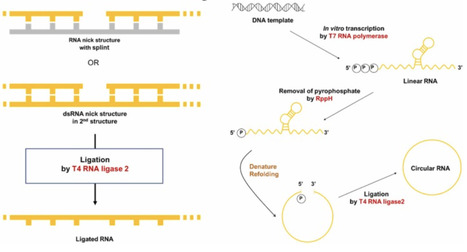
Circular RNA (circRNA) is a covalently closed RNA molecule in which the 5′ and 3′ ends are joined. CircRNAs can be generated via RNA circularization, a process that links the termini of linear RNA. The most common in vitro method for RNA circularization is the ribozyme-based permuted introns and exons (PIE) system. However, circRNAs produced by the PIE method retain partial exogenous exon sequences, potentially leading to immunogenicity issues. In this study, we developed an alternative approach that exploits the secondary structure of linear RNA and enzymatic ligation to synthesize circRNA in vitro for targeted gene expression. We first predicted RNA secondary structures and designed linear RNA molecules to form a terminal nick structure. Using T4 RNA ligase 2 (T4 Rnl2), we sealed the nick to connect the RNA ends. This ligase-mediated in vitro circularization achieved high efficiency and enabled functional expression of the encoded target gene. While ligase-based splint-free circularization has been described for small RNA circles, reports demonstrating functional protein expression from such constructs remain limited. This ligase-mediated RNA circularization approach should be an efficient alternative for production of circular RNA. This ligase-mediated, secondary-structure–guided, splint-free strategy using T4 RNA ligase 2 represents an efficient alternative for circRNA production, enabling both high-yield synthesis and validated gene expression in mammalian cells.
|
|
Scooped by
?
December 13, 7:12 PM
|
Antimicrobial resistance (AMR) poses a pressing global health challenge in the 21st century. The rapid increase and prevalence of multidrug-resistant bacteria will require novel approaches to develop new antibiotics. Major advances in nucleic acid-based therapeutics, particularly antisense technologies, could be one solution for developing precision antibiotics. The selectivity and specificity in the drug design of antibacterial antisense oligomers (ASOs) allows precise gene-specific silencing and ultimately enables targeting of currently undruggable gene products. Our goal here is to comprehensively review the advances in asobiotics (antisense oligomer biotics) leading to therapeutic success, including: modifications in the nucleic acid backbone of ASOs which have improved their properties and advances in delivery. We will discuss utilization of ASOs against several pathogens, strategies to overcome resistance, and finally future scenarios and prospects for asobiotics as pathogen-specific therapy in the clinic.
|
|
Scooped by
?
December 13, 7:00 PM
|
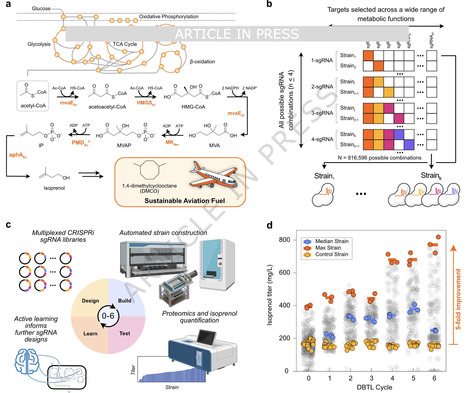
Advances in genome engineering have improved our ability to perturb microbial metabolic networks, yet bioproduction campaigns often struggle with parsing complex metabolic datasets to efficiently enhance product titers. We address this challenge by coupling laboratory automation with machine learning to systematically optimize the production of isoprenol, a sustainable aviation fuel precursor, in Pseudomonas putida. The simultaneous downregulation through CRISPR interference of combinations of up to four gene targets, guided by machine learning, permitted us to increase isoprenol titer 5-fold in six consecutive design-build-test-learn cycles. Moreover, machine learning enabled us to swiftly explore a vast experimental design space of 800,000 possible combinations by strategically recommending approximately 400 priority constructs. High-throughput proteomics allowed us to validate CRISPRi downregulation and identify biological mechanisms driving production increases. Our work demonstrates that ML-driven automated design-build-test-learn cycles, when combined with rigorous data validation, can rapidly enhance titers without specific biological knowledge, suggesting that it can be applied to any host, product, or pathway. Laboratory automation, machine learning, and metabolic engineering may be combined to quickly and efficiently build productive microbial strains. Here the authors used these techniques in P. putida to boost isoprenol titers 5-fold over six DBTL cycles while sampling a reduced design space.
|
|
Scooped by
?
December 13, 5:14 PM
|
Adenine base editors (ABEs) are powerful tools for gene therapy. However, efficient version of ABEs (e.g. ABE8e) always induce excessive bystander and off-target editing events and are large in size, hindering their potential in clinical disease treatment. Here, we develop a pre-trained Protein-Nucleic Acid Constrained Language Model to design ABE8e with high activity, reduced editing window and decreased size. By further engineering, the smallest ABE8e- PNLM-pcABE- with a 27% size reduction, exhibits high activity, precise 3-nt editing window, and reduced off-target events near background level in HEK293T cells. Compared to ABE8e, PNLM-pcABE has up to 133.5-fold precision improvement in pathogenic mutation correction. By PNLM-pcABE, the albino mouse model carrying desired base mutation is nearly 100% obtained via zygotes microinjection and the expression of PCSK9 substantially decreases in mice receiving in vivo delivery with lipid nanoparticle (LNP), indicating their great potential in gene therapy and disease modeling. Adenine base editors (ABEs) are powerful tools for gene therapy, though efficient versions of ABEs often induce excessive undesired editing events, limiting clinical application. Here, authors developed a Language Model to design a more precise ABE that can correct pathogenic point mutations and enable gene therapy in vivo.
|
|
Scooped by
?
December 13, 5:03 PM
|
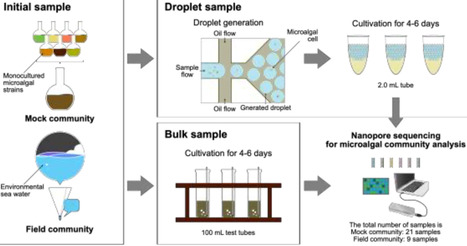
Efficient screening of microalgae is critical for their research and industrial applications. However, conventional methods of microalgae screening are often considered inefficient and are applicable to a narrow range of species. We conducted a proof-of-concept study on the applicability of a water-in-oil droplet (WODL)-based microfluidic system to high-throughput screening of microalgae and effect of microfluidic droplet cultivation on the species diversity within microalgal samples. First, we confirmed that microalgae with diverse morphologies could be successfully cultured within droplets. Second, we conducted culture tests using a mock community derived from monocultured microalgal strains and a field community derived from the natural environment. Subsequent long-read metabarcoding targeting the 16 S rRNA gene, followed by diversity analysis, revealed that droplet culture is more effective than bulk mixed-species culture in maintaining species diversity. Our results lay the groundwork for the future development of WODL-based high-throughput screening systems, allowing access to a richer microalgal biodiversity.
|
|
Scooped by
?
December 13, 4:56 PM
|
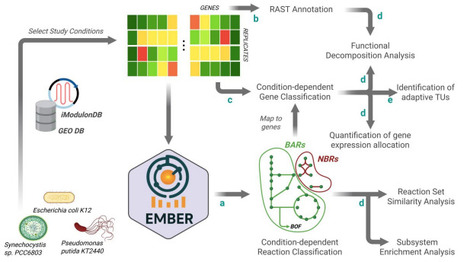
Microorganisms maintain resilience in fluctuating environments by operating close to a multi–optimality state, balancing growth rate and adaptability. This trade–off dictates bacterial resilience and often complicates metabolic engineering efforts. Addressing it requires identifying specific pathways responsible for diverting metabolic resources away from desired production goals. For this purpose, we introduce EMBER (Exploration of Metabolic trade–offs Based on the mapping of Expression patterns to Reactions), a novel Genome–scale Metabolic Model (GEM) contextualization approach. EMBER integrates transcriptomic data and flux analysis to computationally distinguish between growth–associated reactions (BARs) and adaptive, non–biomass reactions (NBRs). We applied this framework to analyze the metabolic architectures of three diverse and biotechnologically relevant organisms—P. putida, E. coli, and Synechocystis—across various environmental conditions. We revealed marked variability in adaptive resource allocation, with the heterotrophs dedicating substantially more active genes to NBRs (up to 31%) than the photoautotroph Synechocystis (17.5%). Functional analysis showed that BARs consistently supported core metabolism, while NBRs encoded context–specific adaptive functions aligned with the native environment of each organism. Analysis of NBR gene expression variability further suggested that P. putida relies predominantly on Bet–Hedging strategies, whereas E. coli employs more regulated Responsive Switching mechanisms. Overall, EMBER offers a powerful systems biology tool to quantify and functionally interpret metabolic heterogeneity. This systematic identification of NBRs will facilitate precise metabolic engineering efforts via reducing unnecessary fitness costs or harnessing the population heterogeneity for deploying complex biotechnological tasks.
|
|
Scooped by
?
December 13, 4:37 PM
|
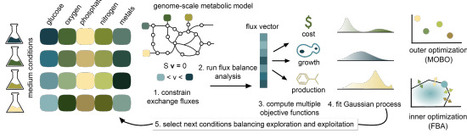
The optimization of culture media is critical for improving the efficiency and cost of cellular production systems. Traditional approaches often rely on extensive experimental trials or statistical methods, which can be both costly and time-consuming. Here, we present gsMOBO, a computational approach for media design that integrates genome-scale metabolic models with multiobjective optimization. Our method employs Flux Balance Analysis coupled with a top-layer Bayesian optimization routine for efficient exploration and optimization of nutrient combinations across high-dimensional spaces. We show that our method finds optimal medium formulations along a Pareto front balancing growth, production and cost of medium components. We illustrate the approach in models of Escherichia coli engineered to produce antibody fragments, as well as Bacillus subtilis strains that synthesize cyclic lipopeptides. Our results show that gsMOBO identifies media compositions and Pareto-optimal trade-offs consistent with prior experimental work. Our method provides a broadly applicable tool for the design of cost-effective and productive culture media, and offers a route to accelerate medium development in biomanufacturing.
|
|
Scooped by
?
December 13, 4:20 PM
|
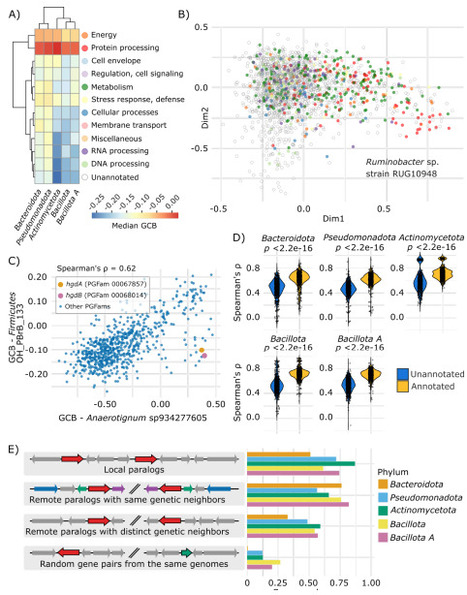
The usage of synonymous codons varies along the genome, with strong biases in conserved and highly expressed genes that are optimized for translation. The extent of codon usage adaptation across genes and co-adaptation between genes, as well as the influence of gene function on these patterns, remain important open questions. Here, we show that codon usage is highly non-random in most bacterial genes, with at least ~20-46% of the gene families presenting synchronized codon usage evolution. We show that co-adapting genes are co-expressed, co-regulated, metabolically connected, and functionally associated. Codon usage adaptations of key marker genes highlight differences in their expression context between microbes with alternative ecological strategies. This underappreciated regulatory dimension has important implications for function discovery and engineering.
|
|
Scooped by
?
December 13, 4:08 PM
|
Blood cancers such as leukemia, lymphoma, and myeloma remain refractory in many patients due to immune escape, antigen heterogeneity, and therapy‑related toxicities. To address these challenges, we review recent strategies that harness CRISPR‑engineered gut commensals as precision “living therapeutics” to modulate host immunity and directly target malignant clones. We frame this review around three principal themes: (1) mechanistic strategies whereby CRISPR-engineered commensals modulate host immunity and directly antagonize malignant clones; (2) the enabling technologies and delivery/containment platforms, CRISPR variants, phage/LNP delivery, genetic circuits and biocontainment, that make living therapeutics feasible; and (3) translational progress, outstanding technical and safety barriers, and ethical/regulatory challenges that must be addressed for clinical deployment. To illustrate these themes, we discuss three concrete therapeutic modalities: engineered microbial secretion of immunomodulators, targeted delivery of tumor-lytic payloads, and engineered production of anticancer metabolites, and how these are enabled by contemporary CRISPR and synthetic-biology toolkits. Selected preclinical models report substantial antitumor effects, often >60% tumor reduction in rodent studies, and restoration of CAR-T cell function in controlled settings; however, effect sizes vary across models, and human translation remains unproven. We also analyze key technical barriers, strain stability, biocontainment, off‑target effects, and propose solutions, including auxotrophic kill-switches and AI‑guided strain optimization. Finally, we outline future directions, from in situ phage delivery to multi‑omics–driven patient stratification. CRISPR‑microbiome editing represents a paradigm shift in hematologic oncology, offering localized, sustained therapy with reduced systemic toxicity.
|
|
Scooped by
?
December 13, 3:43 PM
|
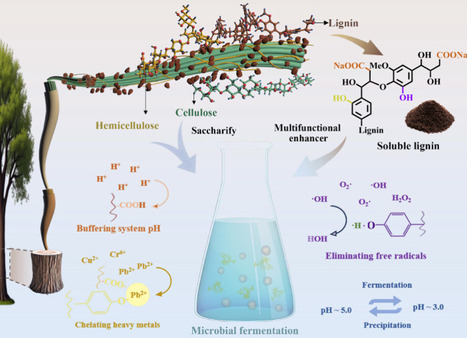
Transforming lignocellulose into high-value biochemicals via microbial fermentation is critical for valorizing agricultural/forestry residues and advancing sustainable biomanufacturing. However, fermentation efficiency is hindered by environmental stressors. Herein, soluble lignin was prepared by alkaline dissolution and dialysis of enzymatic hydrolysis lignin and its effects on the above three fermentations were systematically investigated. Results showed that soluble lignin buffered fermentation pH via carboxyl protonation, scavenged ROS through phenolic hydroxyl groups to alleviate microbial inactivation, and chelated heavy metals (e.g., Pb2+, Cd2+) via phenolic hydroxyl/carboxyl coordination to reduce toxicity. Notably, it exhibited pH-responsive recyclability: dissolving at neutral pH (initial fermentation) and precipitating at acidic pH (fermentation end), enabling centrifugal recovery and reuse for ≥10 cycles with retained performance. This study demonstrates that soluble lignin universally enhances multiple microbial fermentations via physicochemical synergies, providing a theoretical basis for optimizing lignocellulose-based fermentation and new avenues for high-value lignin utilization in biomanufacturing.
|
|
Scooped by
?
December 13, 3:29 PM
|
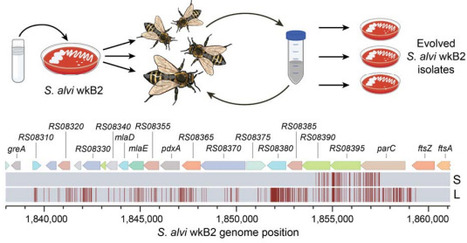
Sexual recombination and horizontal gene transfer are expected to improve the survival of host-associated bacteria that face colonization bottlenecks and intense competition. We serendipitously observed recombination between Snodgrassella alvi strains within bees, which led us to discover that this core member of the bee gut microbiota is naturally competent, including under laboratory conditions. High rates of gene transfer via DNA release and uptake by S. alvi have implications for how it has evolved in managed hives and suggest opportunities for using in situ microbiome engineering to protect bee health.
|
|
|
Scooped by
?
December 13, 7:28 PM
|
Squared-off boxes with illustrations represent data structures (light grey background) or analyses (white background). Round-cornered boxes represent specific analytical tools, categorized as follows: green, functionality implemented in scikit-bio; blue, functionality offered by external Python libraries that can be used in conjunction with scikit-bio within the Python framework—for example, a distance matrix generated by scikit-bio can be input into SciPy for hierarchical clustering, scikit-learn for k–nearest neighbors classification or umap-learn for UMAP embedding; yellow, functionality provided by non-Python programs that can interact with scikit-bio through file input and output (I/O). For example, scikit-bio can read a phylogenetic tree built by RAxML or a multiple sequence alignment generated by MAFFT.
|
|
Scooped by
?
December 13, 7:23 PM
|
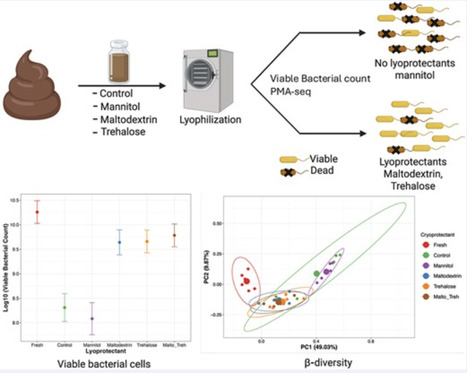
Fecal microbiota transplantation (FMT) is a promising approach for restoring gut microbial balance in both humans and animals. However, the logistical limitations of transplanting fresh fecal samples have increased interest in freeze-dried (lyophilized) fecal material as a transplant inoculum. While lyophilization facilitates storage, it can compromise bacterial viability, which is essential for FMT effectiveness. Lyoprotectants are often used to protect bacterial cultures during freeze-drying, but their effects on a complex microbial community remain unclear, as they may preferentially preserve some taxa over others. This study investigated the impact of four lyoprotectants—mannitol, maltodextrin, trehalose, and a maltodextrin-trehalose mixture—on bacterial viability and community structure in pig fecal samples post-lyophilization. Propidium monoazide (PMA) treatment combined with 16S rRNA sequencing (PMAseq) was used to differentiate viable from non-viable bacteria. In the total community (without PMA), microbial profiles appeared similar across treatment groups. However, when focusing on the viable community (PMA-treated), lyoprotectant choice significantly influenced the post-lyophilization community composition. Gram-negative bacteria were especially susceptible to viability loss due to lyophilization. Trehalose and maltodextrin preserved bacterial viability and community structure more effectively than mannitol. Mannitol-treated samples had reduced viable bacterial cells and altered community composition, while trehalose and maltodextrin better maintained diversity and structure of the viable (PMA-treated) communities. Taken together, lyoprotectants have differential effects on microbial composition during lyophilization. Among those tested, trehalose and maltodextrin best preserved both viability and community structure, making them promising candidates for FMT applications. Future research should explore optimizing lyoprotectant formulations to enhance microbiome stability and functional outcomes.
|
|
Scooped by
?
December 13, 7:15 PM
|
World agriculture depends in part on the crop-associated microbiome for improved plant growth, health, and productivity. In particular, endophytic fungi (EF) with plant growth–promoting activities fulfill some of these roles and are central as bioinoculant agents. In the case of arbuscular mycorrhizal fungi (AMF), they form a symbiosis with their host plants, enhancing the uptake of water, phosphorus, nitrogen, and other micronutrients, while the plants provide them with photosynthates. This work reviews the differences in the colonization of internal plant niches between these beneficial fungi, as well as other distinctive ecological traits. It also explores mechanisms of seedborne vertical transmission in AMF and their classification. Genomic and transcriptomic advances in fungal endophytes are highlighted, shedding light on genes and expression profiles that define their lifestyle and plant associations. In addition, recent studies on their abilities to promote plant growth are analyzed, especially focusing on Trichoderma spp., Epichloë spp., Serendipita indica (formerly Piriformospora indica), and entomopathogens like Beauveria spp. and Metarhizium spp. Finally, the multiple interactions among EF, AMF, and other members of the plant microbiome—notably plant growth-promoting bacteria (PGPB)—are discussed, emphasizing how these organisms synergistically benefit the host. A deeper understanding of these fungi and their plant-beneficial effects should facilitate commercialization and help farmers achieve sustainable production, especially under challenges posed by global climate change.
|
|
Scooped by
?
December 13, 7:04 PM
|
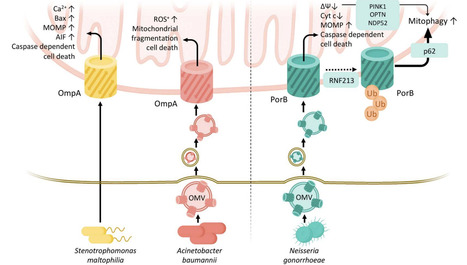
Gram-negative bacteria are equipped with a unique cell envelope structure that includes an outer membrane populated by diverse outer membrane proteins (OMPs). These OMPs are not only essential for bacterial survival, mediating critical functions such as nutrient transport, antibiotic resistance, and structural integrity, but they also play pivotal roles as virulence factors during host-pathogen interactions. Recent research highlights the ability of OMPs to manipulate host cellular processes, often targeting mitochondria to induce cell death or modulate immune responses. This review explores the multifunctional roles of bacterial OMPs, emphasizing their structural features, biogenesis, and pathogenic mechanisms. Furthermore, it delves into how bacterial OMPs exploit host cell machinery, particularly mitochondria, to promote infection, as well as their potential as targets for innovative antimicrobial strategies. Specifically, this review focuses on β-barrel OMPs that reach host mitochondria, detailing their delivery routes and mechanisms of organelle manipulation, while excluding non-β-barrel toxins and secretion-system effectors, to provide a defined perspective on mitochondria-targeting OMP virulence mechanisms. omv
|
|
Scooped by
?
December 13, 6:51 PM
|
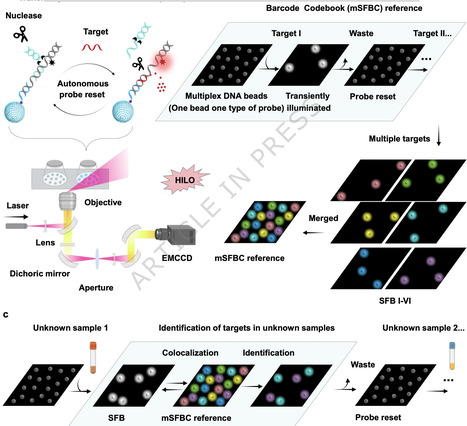
Multiplexed nucleic-acid detection is essential for molecular diagnostics and spatial genomics, but conventional fluorescence methods are often limited by spectral overlap, nonspecific signals, and restricted encoding capacity. We present a spatial fluorescence barcode (SFB) platform based on transiently luminescent DNA beads (TLDBs) that enables single-color, high-plex readout. In this method, targets are encoded through the spatial arrangement of DNA-functionalized beads, eliminating the need for multicolor labeling or spectral unmixing. Target recognition is achieved through toehold-mediated strand displacement, and built-in nucleases enable autonomous enzymatic resetting for repeated use of probes. The system employs monochromatic spatial encoding, decoupling encoding capacity from spectral channels, and features a simplified probe design and decoding workflow. Self-resetting probes not only streamline the encoding process but also enhance practicality by allowing repeated assays without the need to re-prepare costly probe combinations. We demonstrate robust detection of pathogen-derived nucleic acids in infected blood and cancer-associated microRNAs in tissue samples, validating the platform’s clinical applicability. Compared to existing barcoding strategies, SFB integrates monochromatic spatial encoding, simplified design, and autonomous reusability, offering a practical, scalable, and cost-effective solution for high-throughput nucleic acid analysis. Fluorescence methods of multiplexed nucleic-acid detection are often limited by spectral overlap, nonspecific signals, and encoding capacity. Here the authors design a spatial fluorescence barcode platform which uses DNA beads for reusable multiplexed pathogen and cancer biomarker detection.
|
|
Scooped by
?
December 13, 5:08 PM
|
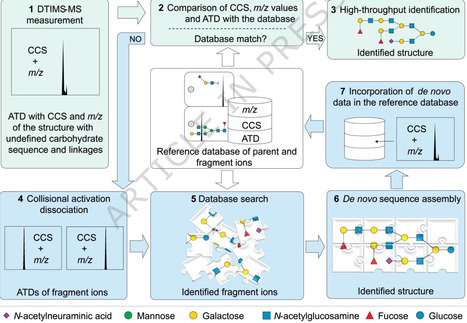
It is essential to determine glycan structures in complex biological samples to understand their biology and exploit their diagnostics, therapeutics and nutraceuticals potential. An unresolved analytical challenge is the identification of isomeric glycan structures in complex biological samples. Ion mobility (IM) combined with MS enables separation of isomeric glycans and identification by comparing their intrinsic collision cross section (CCS) values with similar data of synthetic standards. To identify glycans without the need to synthesize all biologically occurring glycans, we describe here an IM-MS de novo sequencing method based on fragment identification and sequence assembly. CCS values of additional fragments from glycans in biological samples result in a self-expanding reference database, gradually facilitating the sequencing of glycans of increasing complexity and expanding the database from an initial 19 standards to 332 unique entries. The methodology is employed to determine structures of human milk oligosaccharides and N-glycans of biotherapeutics. This study introduces an ion mobility–mass spectrometry method for de novo glycan structure identification, using a self-expanding database that reduces reliance on synthetic reference standards.
|
|
Scooped by
?
December 13, 4:59 PM
|
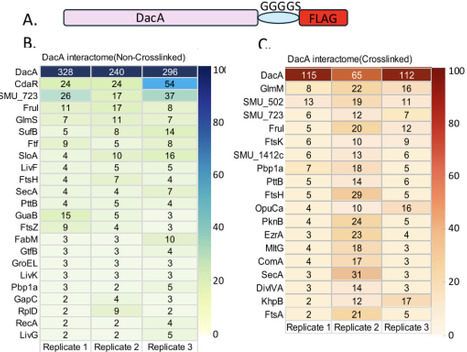
Diadenylate cyclase (DacA) synthesizes the second messenger cyclic di-AMP (c-di-AMP), which regulates essential cellular processes across many Gram-positive and select Gram-negative bacteria/archaeal lineages. Although DacA is known to interact with regulators such as GlmM and CdaR, the breadth and functional relevance of its interactome remains poorly defined. Our study seeks to identify novel protein-protein interactions to further elucidate their unknown regulatory mechanisms and cellular roles. Using Streptococcus mutans as a model, we engineered a Flag-tagged strain (DacA-FLAG) and then performed co-immunoprecipitation under non-crosslinked and crosslinked conditions followed by mass spectrometry. We identified 22 candidates interacting proteins in non-crosslinked samples, 18 in crosslinked samples, and 6 shared between conditions. Selected partners were validated in vivo using split luciferase complementation. Notably, SMU_723 emerged as a key binding partner. AlphaFold-guided modeling predicated a direct DacA and SMU_723 interaction interface involving threonine 147, glutamine 148, and threonine 149 in DacA . Site-directed mutagenesis of these residues impaired binding, confirming their critical role. An SMU_723 deletion phenocopied a dacA deletion strain, sharing prolonged lag phase, aberrant cell morphology, reduced acid production and acid tolerance, impaired sorbitol metabolism, decreased colonization in a Drosophila model, and delayed growth upon calcium stimulation. These shared phenotypes suggest a functional and possibly regulatory link between DacA and SMU_723. Given SMU_723 sequence homology consistent with a calcium transporter, these data suggest that the DacA–723 interaction contributes to calcium homeostasis and/or calcium-responsive signaling.
|
|
Scooped by
?
December 13, 4:43 PM
|
Metabolic modeling enables the prediction of functional capabilities in organisms and microbial communities from genomic data. However, current workflows for genome-scale metabolic model (GEM) reconstruction and contextualization remain time-consuming and technically demanding, particularly when integrating multi-omics data or deriving community-level models from taxonomic profiles. Although recent advances have improved automation and omics integration, challenges persist in incorporating heterogeneous datasets such as single-cell RNA sequencing and in interpreting microbiomes from 16S rRNA data. We present a computational tool for rapid, automated GEM generation with integrated support for contextualization using transcriptomic and single-cell omics data. The platform also enables the construction of core consortium metabolic models from 16S rRNA profiles, facilitating systems-level interpretation of both single-organism and community-scale datasets. This streamlined pipeline offers a scalable solution for microbiome research, including population heterogeneity analysis and metabolic engineering. Its utility has been demonstrated by exploring phenotypic heterogeneity within Bacillus subtilis populations and identifying metabolic interactions among members of a cyanobacteria-enriched microbial consortium.
|
|
Scooped by
?
December 13, 4:27 PM
|
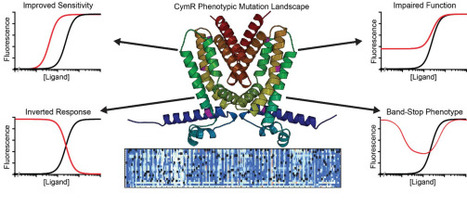
CymR is a TetR-family transcriptional repressor that recognizes a well-defined operator sequence in the promoter PcymRC. The native ligand cumate and several structurally related aromatic acids bind at an allosteric site and induce a conformational change in CymR, resulting in release from the DNA operator and de-repression of the promoter. The amino acid residues that contribute to these core functions have not been mapped, nor has the protein been subjected to extensive mutagenesis to modify its function. Here, for the first time, we integrate Deep Mutational Scanning (DMS) with Growth-based Quantitative Sequencing (GROQ-Seq) to evaluate a comprehensive phenotypic landscape of CymR variants, including single amino acid insertions and deletions. We measure this library across a concentration gradient of small molecule inducers to construct an induction curve for all library members. From this analysis, we identify amino acids throughout the protein that are essential for repressor function and discover several mutations that improve the sensitivity of CymR to the ligand perillic acid. In addition, rarely investigated insertion mutants are revealed to be a key driver of novel phenotypes, including several regions of CymR where insertions result in an inverted phenotype and the isolation of variants exhibiting an unusual band-stop phenotype.
|
|
Scooped by
?
December 13, 4:15 PM
|
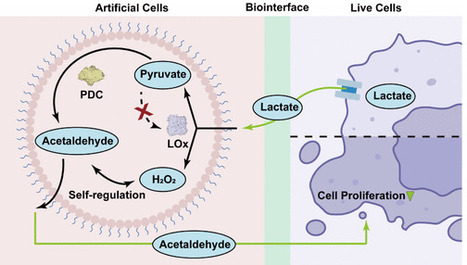
Artificial cells (ACs) offer a powerful platform to reprogram metabolic signaling in complex tissue environments by replicating key biological functions without the full complexity of living cells. However, achieving autonomous metabolite exchange and stable integration with living tissues remains a major challenge. Here, we report the development of proteinosome-based ACs equipped with a minimal metabolism to mediate bidirectional communication with glycolytic tumor cells. These tumors accumulate lactate, a metabolic byproduct that promotes immunosuppression and metastasis. Although lactate oxidase (LOx) can degrade lactate, its oxidation product, pyruvate, may inadvertently fuel tumor growth. To overcome this limitation, we engineered dual-processor ACs coencapsulating LOx and pyruvate decarboxylase (PDC), enabling selective conversion of lactate into cytotoxic acetaldehyde while suppressing pyruvate and hydrogen peroxide accumulation. These ACs demonstrate sustained catalytic activity, maintain reactive oxygen species homeostasis, and remain functional when integrated in 3D tumor spheroids. Crucially, they engage in autonomous, bidirectional metabolite exchange, preferentially with cancer cells over normal cells, dynamically rewiring important metabolites of the tumor microenvironment and suppressing cell viability. This work establishes synthetic metabolic biointerfaces as programmable actuators capable of reshaping pathological signaling in cancer tissues.
|
|
Scooped by
?
December 13, 3:46 PM
|
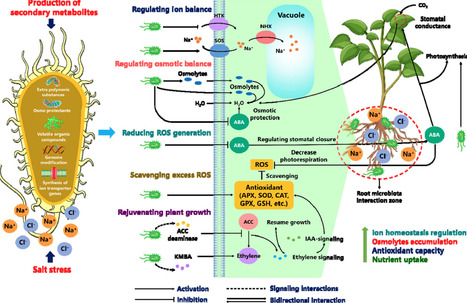
Soil salinity is a major abiotic stress limiting crop productivity and long-term soil health by inducing osmotic and ionic imbalances, disrupting water and nutrient uptake. Traditional mitigation methods like soil leaching, chemical amendments, and breeding salt-tolerant crops offer limited success due to high costs and environmental concerns. Therefore, attention has shifted toward utilizing the soil microbiome, including plant growth-promoting rhizobacteria, mycorrhizal fungi, and halotolerant microbes, which support through various physiological mechanisms. These include phytohormone modulation, antioxidant activation, nutrient acquisition and biofilm formation. Unlike earlier studies, this review provides a comprehensive synthesis of microbiome-mediated physiological mechanisms that enhance plant salt tolerance. Moreover, it explores how salinity affects microbial communities that disturbs soil nutrient cycles. The outcomes emphasize that there is a pressing need to transition from chemical-based methods to microbe-assisted approaches. Future research should prioritize the development of location-specific microbial consortia and long-term field trials assessing soil-microbe-plant interactions under salinity.
|
|
Scooped by
?
December 13, 3:35 PM
|
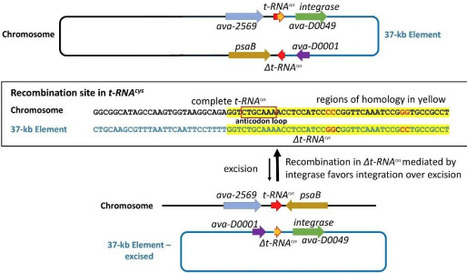
Anabaena (aka Trichormus) variabilis ATCC 29413 strain FD is a filamentous, heterocyst-forming cyanobacterium with a 6.36 Mb chromosome, three circular plasmids, A (366 kb), B (35.8 kb), C (301 kb), and a 37-kb excision element. This 37-kb element in A. variabilis ATCC 29413 strain FD was integrated into the tRNAcys gene but was absent in the very closely related strains A. variabilis FSR and PNB. The 37-kb element was also excised as a circular molecule at a very low frequency compared to the integrated form. The 37-kb element has a partial copy of tRNAcys; therefore, integration produced a functional but genetically distinct copy of tRNAcys. Integration and excision are likely mediated by a putative integrase with a tyrosine recombinase domain encoded within the element, possibly using the duplicated copies of the 7-bp anticodon of the tRNAcys as a recombination site. Like many other bacterial elements, the function of this element is unknown, but, like other well-characterized excision elements in heterocystous cyanobacteria, it does not appear to be important for survival.
|