 Your new post is loading...

|
Scooped by
?
Today, 1:17 AM
|
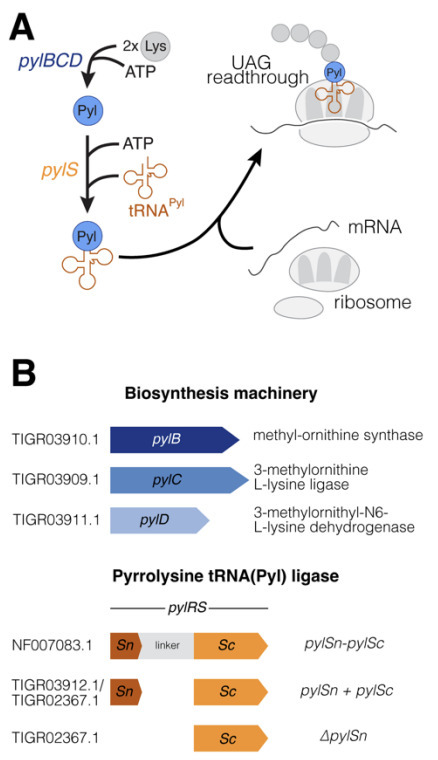
Natural genetic code expansion is a phenomenon wherein an additional amino acid is encoded by a stop codon. These non-standard amino acids are beneficial as they facilitate novel biochemical reactions. However, code expansion leads to ambiguity at the recoded stop codon, which can either be read through or terminated. Pyrrolysine (Pyl) is encoded by the amber codon (TAG/UAG) and is widespread in archaea, where it is required for methylamine-mediated methanogenesis, an environmentally important metabolism. Mechanisms to conditionally suppress the amber stop codon for Pyl installation during protein synthesis have not been identified. Using the model methanogen, Methanosarcina acetivorans, we demonstrate that Pyl-encoding archaea maintain an ambiguous genetic code wherein UAG encodes dual meaning as stop and Pyl. Our data suggest that expression of Pyl biosynthesis and incorporation genes is tuned to the cellular demand for Pyl, which allows these archaea to navigate ambiguous stop decoding in response to environmental cues.
|
|
Scooped by
?
Today, 12:29 AM
|
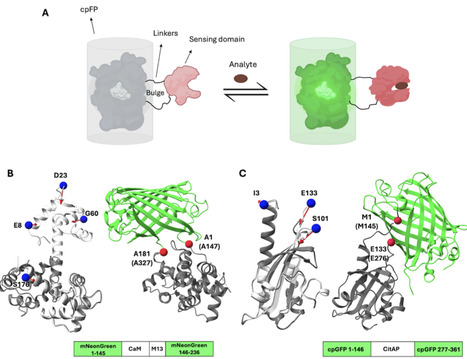
Proteins exhibit remarkable conformational flexibility, enabling precise functional regulation through allostery. A key application of allostery is in the design of protein-based sensors, which detect environmental changes—such as ligand binding or post-translational modifications—and convert these cues into measurable signals (e.g., fluorescence). Here, we investigate a series of ligand-binding proteins that serve as sensing domains in direct-response fluorescent biosensors, wherein ligand binding enhances fluorescence output. We employ a multiple force application approach which we term Multiply Perturbed Response (MPR) to identify “hot spot” residues that drive the conformational transition from an apo (inactive/OFF) to a holo (active/ON) state. We first present two efficient computational approaches to determine residues and forces that maximize the overlap of the observed conformational change. We then determine the overlap maximizer residues for up to five force insertion locations, and we compare them with actual insertion sites used in existing biosensors. Our analysis shows that the allosteric residues identified by MPR coincide with the fluorescent-protein insertion sites that were mapped experimentally through extensive trial-and-error. This work enhances the utility of linear response theory-based methods in uncovering multiple functionally significant regions that trigger a known conformational change. While the validity of the harmonic approximation in anharmonic conformational transitions needs additional validation, MPR gives a good starting point to explore allosteric sites. The approach might prove useful not only in the design of biosensors, but may also find applications in offering physics-based collective variables in mapping conformational transition pathways of proteins.
|
|
Scooped by
?
June 11, 11:50 PM
|
Microbial communities, complex ecological networks crucial for human and planetary health, remain poorly understood in terms of the quantitative principles governing their composition, assembly, and function. Dynamic modeling using ordinary differential equations (ODEs) is a powerful framework for understanding and predicting microbiome behaviors. However, developing reliable ODE models is severely hampered by their nonlinear nature and the presence of significant challenges, particularly critical issues related to identifiability. Here, we address the identification problem in dynamic microbial community models by proposing an integrated methodology to tackle key challenges. Focusing on nonlinear ODE-based models, we examine four critical pitfalls: identifiability issues (structural and practical), unstable dynamics (potentially leading to numerical blow-up), underfitting (convergence to suboptimal solutions), and overfitting (fitting noise rather than signal). These pitfalls yield unreliable parameter estimates, unrealistic model behavior, and poor generalization. Our study presents a comprehensive workflow incorporating structural and practical identifiability analysis, robust global optimization for calibration, stability checks, and rigorous predictive power assessment. The methodology’s effectiveness and versatility in mitigating these pitfalls are demonstrated through case studies of increasing complexity, paving the way for more reliable and mechanistically insightful models of microbial communities. https://doi.org/10.5281/zenodo.15309438
|
|
Scooped by
?
June 11, 11:13 PM
|
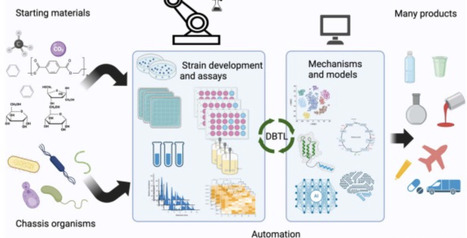
Microbial production of target molecules has advanced significantly in recent years driven by innovations in enzyme engineering, DNA synthesis, and genomic editing. However, to access the massive potential of microbial production, a vast parametric space remains to be investigated to optimize these biobased processes for a robust bioeconomy. Here, we review the current state of the art, some key challenges and possible solutions. We see a critical role of automation, high-throughput technologies, self-driving and cloud labs, and data management to enable Artificial Intelligence/Machine Learning and mechanistic models to overcome the design space challenges and accelerate the development of novel bio-based solutions. Accurate models will expedite the development and scale-up of engineered microbes for a range of final products from many starting materials.
|
|
Scooped by
?
June 11, 10:58 PM
|
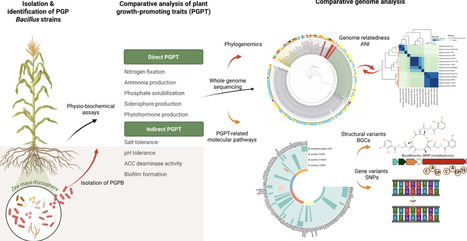
Bacillus species are among the most promising plant growth-promoting bacteria (PGPR) due to their adaptability to various environmental niches and extensive biosynthetic capabilities. Despite the available data on the PGP-traits of Bacillus, the genetic basis underlying their beneficial effects remains largely unexplored. In this study, a comparative genomic analysis of three B. pumilus and one B. pseudomycoides strains, isolated from the maize rhizosphere, is presented to elucidate the molecular mechanisms behind their PGP-traits. All strains exhibited multiple PGP-traits, including phosphate solubilization, phytohormone and siderophore production, growth in nitrogen-free medium, stress tolerance, and biofilm formation. Phylogenomic analysis revealed that plant-associated strains have higher genetic similarity, emphasizing niche-specific evolution. Genome analyses revealed strain-and species-specific adaptations, particularly in relation to nutrient acquisition and abiotic stress response mechanisms. B. pumilus strains encoded alternative sigma factors (SigB, SigM, SigW) enabling enhanced salt tolerance, whereas B. pseudomycoides lacked this system and relied on conventional osmoprotective strategies. The strains utilized different tryptophan-dependent (IAN, IAM or IPyA) pathways for auxin biosynthesis and differed in phosphate solubilization ability, which can be attributed to upstream and missense variants in genes affecting acid metabolism (gltA, acnA, acnB, citM, and citS) and phosphatase (phoA) activity. Iron uptake via bacillibactin-siderophores was exclusive to B. pumilus. The inability of the B. pseudomycoides strain to acquire iron was associated with structural variants (absence of bsaA gene) within the bacillibactin biosynthetic gene cluster. This work provides new insights into the molecular basis of PGP traits in Bacillus and supports the development of Bacillus-based bioinoculants for sustainable agriculture.
|
|
Scooped by
?
June 11, 7:29 PM
|
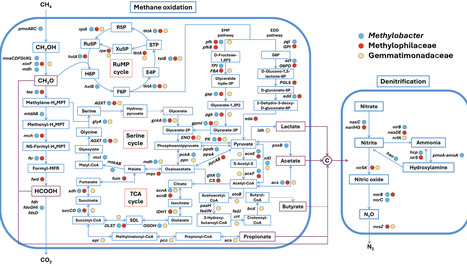
The impact of anaerobic oxidation of methane (AOM) coupled with denitrification on the emission of the denitrification intermediate N2O remains poorly understood. Here, we investigated the influence of AOM coupled with nitrate and nitrite reduction on soil N2O emissions and the associated microbial interactions. We show that AOM coupled with denitrification markedly reduces soil N2O emissions, with the type I methanotroph Methylobacter species collaborating with the Methylophilaceae and Gemmatimonadaceae families to perform key roles. The suppression of N2O emissions by AOM primarily stems from its role in supplying electrons and carbon sources, fueling complete denitrification by associated bacteria. In addition, we uncovered distinct microbial interaction strategies for AOM-coupled nitrate and nitrite reduction. While nitrite reduction necessitates both bacterial cooperation and extracellular electron transfer during its initial stages, nitrate reduction predominantly depends on methanotrophic bacteria alone at the outset. These findings advance our understanding of carbon-nitrogen cycle coupling and underscore AOM’s potential to simultaneously mitigate both CH4 and N2O emissions.
|
|
Scooped by
?
June 11, 6:50 PM
|
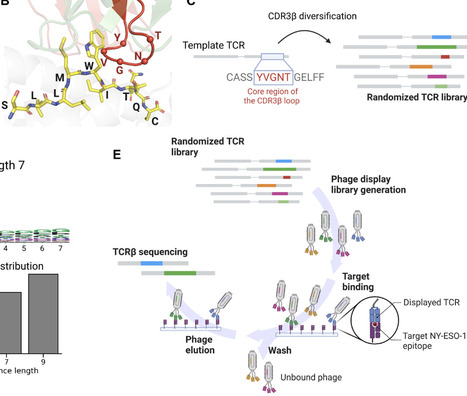
T cells targeting epitopes in infectious diseases or cancer play a central role in spontaneous and therapy-induced immune responses. Epitope recognition is mediated by the binding of the T cell receptor (TCR), and TCRs recognizing clinically relevant epitopes are promising for T cell–based therapies. Starting from a TCR targeting the cancer-testis antigen NY-ESO-1157–165 epitope, we built large phage display libraries of TCRs with randomized complementary determining region 3 of the β chain. The TCR libraries were panned against NY-ESO-1, which enabled us to collect thousands of epitope-specific TCR sequences. Leveraging these data, we trained a machine learning TCR-epitope interaction predictor and identified several epitope-specific TCRs from TCR repertoires. Cellular assays revealed that the predicted TCRs displayed activity toward NY-ESO-1 and no detectable cross-reactivity. Our work demonstrates how display technologies combined with TCR-epitope interaction predictors can effectively leverage large TCR repertoires for TCR discovery.
|
|
Scooped by
?
June 11, 6:20 PM
|
Cas12j-8 is a compact Cas nuclease discovered from the metagenome of giant bacteriophages, consisting of only 717 amino acids and recognizing the ‘5-TTN-3′ protospacer adjacent motif (PAM) sequence. However, its low gene editing efficiency in mammalian cells limits its application in therapeutic gene editing. To address this limitation, structure-guided mutagenesis is employed to replace key negatively charged residues with arginine, strengthening DNA binding. The resulting quintuple mutant, engineered Cas12j-8 (enCas12j-8), demonstrates robust on-target editing efficiency comparable to LbCas12a while maintaining low off-target effects. Cytosine base editors (CBEs) and adenine base editors (ABEs) are developed using enCas12j-8, achieving up to 29.54-fold C-to-T and 36.57-fold A-to-G conversion efficiency compared with the wild-type at the dominated sites, respectively. Notably, enCas12j-8 enables multiplexed editing of three genomic loci simultaneously via a single crRNA array, achieving efficiencies comparable to single-guide approaches. Additionally, enCas12j-8-ABE facilitates the disruption of splice acceptor sites, effectively inducing exon skipping in the SOD1 gene. This strategy holds potential significance for therapeutic genome modulation. These findings establish enCas12j-8 as a versatile, high-precision tool for genome engineering, combining efficient delivery, multiplexing capability, and compatibility with diverse editing modalities.
|
|
Scooped by
?
June 11, 4:28 PM
|
Heterologous expression of polyketide synthase (PKS) genes in Escherichia coli has enabled the production of various valuable natural and synthetic products. However, the limited availability of malonyl-CoA (M-CoA) in E. coli remains a substantial impediment to high-titer polyketide production. Here we address this limitation by disrupting the native M-CoA biosynthetic pathway and introducing an orthogonal pathway comprising a malonate transporter and M-CoA ligase, enabling efficient M-CoA biosynthesis under malonate supplementation. This approach substantially increases M-CoA levels, enhancing fatty acid and polyketide titers while reducing the promiscuous activity of PKSs toward undesired acyl-CoA substrates. Subsequent adaptive laboratory evolution of these strains provides insights into M-CoA regulation and identifies mutations that further boost M-CoA and polyketide production. This strategy improves E. coli as a host for polyketide biosynthesis and advances understanding of M-CoA metabolism in microbial systems. Malonyl-CoA (M-CoA) is essential for polyketide biosynthesis, but its limited availability constrains production. Here the authors engineer and evolve an orthogonal M-CoA pathway in Escherichia coli to improve M-CoA metabolism, increasing M-CoA levels and polyketide yields.
|
|
Scooped by
?
June 11, 1:25 AM
|
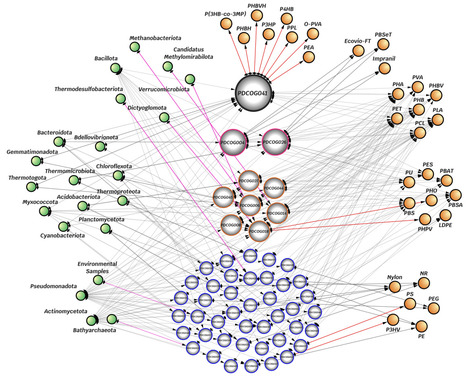
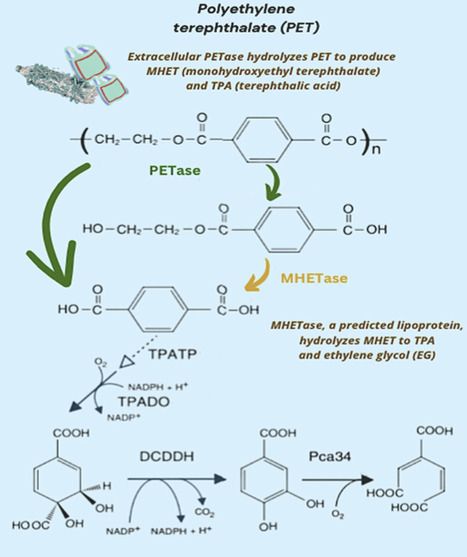
Microplastics and nanoplastics pollution (MNPP) is a major environmental issue due to its small size, long-lasting nature, and potentially harmful impacts on ecosystems and human health. Plastics can be degraded in nature by physical, chemical, and biological processes. Plastic biodegradation is a potential sustainable solution to MNPP, but understanding the fitness of potential plastic-degrading proteins (PPDPs) across environments requires comparative resources. Here, we introduce the Plastic-Degrading Clusters of Orthologous Groups (PDCOGs) database, available at https://phylobone.com/microworld/PDCOG The resource includes 625,616 PPDPs from free-living prokaryotes classified in 51 PDCOGs. The PDCOG database is a robust resource that improves understanding of plastic biodegradation and supports research on global MNPP challenges. Overall, PDPPs represent 3.5% of proteins in prokaryotes and are widely distributed across the Earth, spanning across most prokaryotic species and environments. Nearly all prokaryotic species (>95%) have the potential to biodegrade at least one polymer type.
|
|
Scooped by
?
June 11, 1:06 AM
|
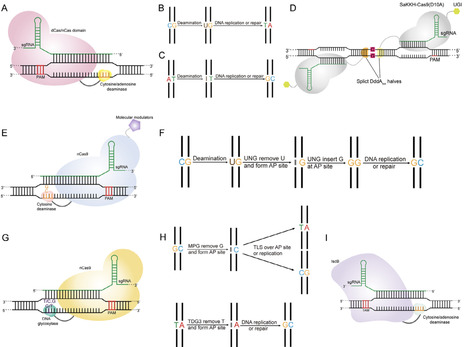
Base editing technology is a novel gene-editing approach derived from the CRISPR/Cas9 system, enabling precise and efficient base conversion. Due to operational simplicity, strong target specificity, high editing efficiency, and minimal editing byproducts, base editing has been widely applied in gene therapy, crop breeding, construction of model organisms, and microbial metabolic engineering. In this review, we systematically summarize the development history and recent advancements of several major base editors, including cytosine base editors (CBEs), adenine base editors (ABEs), CRISPR-free base editors, C-to-G base editors (CGBEs), glycosylase base editors (GBEs), and IscB-derived base editors. Furthermore, we comprehensively summarize optimization strategies for base editors and its application in constructing efficient industrial microorganism, such as Bacillus subtilis, Corynebacterium glutamicum, Saccharomyces cerevisiae, Yarrowia lipolytica, and Aspergillus niger. This review aims to facilitate the broader application of base editing technologies in synthetic biology and accelerate their translational potential.
|
|
Scooped by
?
June 11, 12:57 AM
|
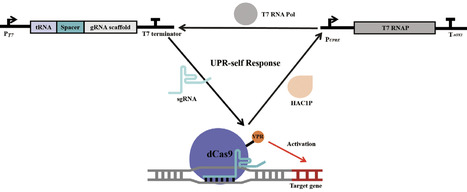
The CRISPR activation (CRISPRa) transcriptional system has become a powerful synthetic biology tool for the regulation of endogenous gene expression, allowing for precise fine-tuning of target genes through the simple modification of sgRNA sequences. In this study, we demonstrate that sgRNAs can be effectively expressed using the T7 transcription system. The insertion of tRNA sequences between PT7 and sgRNAs significantly enhances the efficiency of transcriptional activation. Furthermore, the design of PT7-tRNA-sgRNA arrays facilitates the multiplexed activation of genes. sgRNA expression was regulated by the Tet-on induction system, split-T7 system, and RNA cleavage processing by HH-HDV, resulting in the creation of a Boolean logic gene circuit capable of performing both AND and OR logic operations. Finally, we developed a UPR self-responsive system by utilizing endogenous promoters that are responsive to UPR signals to control the expression of T7 RNAP. This system dynamically regulates the expression of the endogenous HAC1 transcription factor, thus enhancing the secretion of heterologous proteins. The findings from this study highlight the potential of utilizing the T7 transcription system for the construction of genetic circuits, providing a practical toolkit for gene regulation in the industrial Komagataella phaffii strain.
|
|
Scooped by
?
June 11, 12:38 AM
|
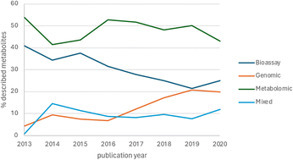
In this review, we analyzed the scientific literature of the period 2013–2023 that reported novel specialized metabolites from the Actinomycetes, one of the most prolific producers of natural products. The discovered metabolites were categorized on the basis of their chemical originality into two groups: variants of known molecules, or original metabolites. In addition, we subdivided the approaches used to discover these metabolites into four categories: bioassay-based screening, genome mining, metabolome mining, or combinations thereof. We present selected examples of the different approaches used and the resulting original metabolites. Finally, we measure the overall trends of discovery in terms of approaches, of the frequency of original metabolites and of the major biosynthetic classes that have been described. Overall, our analysis indicates that new metabolites continue to be discovered from Actinomycetes at a relatively constant rate and that the frequency of original metabolites seems to be approach-independent and relatively constant within the analyzed time period.
|
|
|
Scooped by
?
Today, 12:38 AM
|
Additives such as stabilizers, plasticizers, and fillers are commonly used in relatively small amounts to enhance the structure of plastics. Notably, some of these additives, including moieties of compounds containing nitrogen, phosphorus, and sulfur, are essential for microbial proliferation. Most studies on plastic degradation have primarily focused on the potential of microorganisms to assimilate carbon from plastics to support their growth, a strategy that has yet to yield significant success. However, studies investigating the removal of non‑carbon moieties of additives from plastics, which could weaken their structure and thereby enhance fragmentation, remain largely unexplored. This review highlights the potential of harnessing microbial processes that target the non‑carbon moieties of additives to weaken the structural integrity of plastics. The weakened plastic may then become more accessible to heterotrophic microbes, thereby accelerating its degradation.
|
|
Scooped by
?
Today, 12:10 AM
|
Structural data provides important information on the proteins’ function. Recent development of advanced machine learning and artificial intelligence tools, such as AlphaFold, have led to an explosion of predicted protein structures. However, many of the computed protein models contain unstructured and disordered regions, posing challenges in protein function characterization. Here we present BindUP-Alpha, an upgraded webserver for predicting nucleic acid binding proteins. Our structure-based algorithm utilizes the electrostatic features of the protein surface and other physiochemical and structural properties extracted from the protein sequence. Using a Support Vector Machine (SVM) learning approach, BindUP-Alpha successfully predicts DNA- and RNA-binding proteins from both experimentally solved structures and predicted models. In addition, BindUP-Alpha identifies electrostatic patches on the protein’s surface that represent potential nucleic-acid binding interfaces. BindUP-Alpha is freely accessible at https://bindup.technion.ac.il, providing interactive three-dimensional visualizations and downloadable text-based results.
|
|
Scooped by
?
June 11, 11:37 PM
|
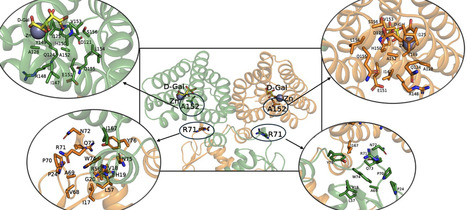
Genetic variations in transcriptional regulators (TRs) of metabolic loci can influence host-bacterial interactions by affecting carbon utilization. Although the metabolism of sugar acids, including D-galactonate, is extensively implicated in the colonization and virulence of enteric bacteria, there has been no investigation on the extent of variations in their pathway-specific TRs. DgoR, the TF of D-galactonate metabolism, is the best-characterized GntR/FadR family sugar acid TFs in enteric bacteria, recognized by the presence of an N-terminal winged helix-turn-helix DNA-binding domain and a C-terminal effector-binding and oligomerization (E-O) domain connected by a linker. Here, we examined 340 Escherichia coli isolates for variations in dgoR and studied their effect on repressor function. Genetic and biochemical studies identified variants with a partial loss of DNA-binding ability (P24L and A152E) and a decreased response to D-galactonate (R71C and P92L). Because the linker residue R71C resulted in a reduced response to D-galactonate and the E-O domain residue A152E led to a DNA binding defect, we performed simulations to probe their altered allosteric behavior. We observed that the correlation patterns, dynamics, and networks of the variants are indeed distinct from the wild type. Importantly, corroborating their repressor function, R71C and A152E variations impacted the growth of natural isolates in D-galactonate. Alignment-based variation detection across all E. coli and Enterobacterales identical protein group data sets revealed less prevalence of these four variations. Collectively, the present study highlights the need for a thorough analysis of the effect of variations in sugar acid TRs on repressor function and their effect on host-bacterial interactions.
|
|
Scooped by
?
June 11, 11:07 PM
|
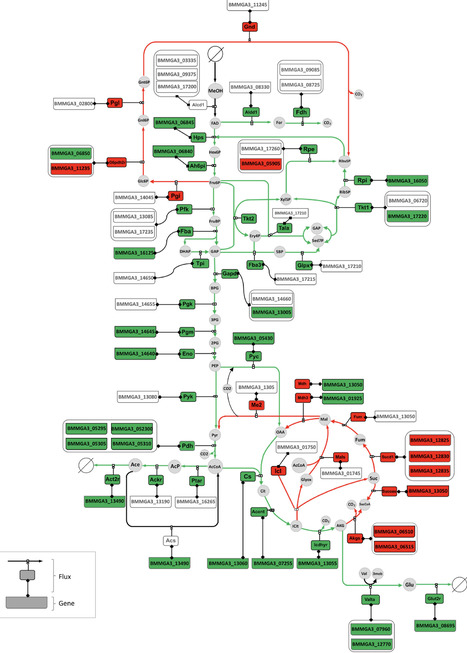
Bacillus methanolicus is the next workhorse in biotechnology using methanol, an alternative and economical one-carbon feedstock that can be obtained directly from carbon dioxide, as both carbon and energy source for the production of value-added chemicals. The wild-type strain B. methanolicus MGA3 naturally overproduces l-glutamate in methanol-based fed-batch fermentations. Here we generated a B. methanolicus strain exhibiting enhanced l-glutamate production capability through induced mutagenesis. To showcase the potential of this mutant strain, further metabolic engineering enabled the production of γ-aminobutyric acid (GABA) directly from l-glutamate during methanol fed-batch fermentations. Using a systems-level analysis, encompassing whole-genome sequencing, RNA sequencing, fluxome analysis and genome-scale metabolic modelling, we were able to elucidate the metabolic and regulatory adaptations that sustain the biosynthesis of these products. The metabolism of the mutant strain specifically evolved to prioritize energy conservation and efficient carbon utilization, culminating in increased product formation. These results and insights provide a foundation for further rational metabolic engineering and bioprocess optimization, enhancing the industrial viability of B. methanolicus for sustainable production of l-glutamate and its derivatives.
|
|
Scooped by
?
June 11, 7:42 PM
|
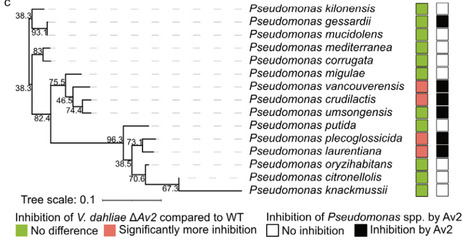
During pathogen attack, plants recruit beneficial microbes in a cry for help to mitigate disease development. Simultaneously, pathogens secrete effectors to promote host colonization through various mechanisms, including targeted host microbiota manipulation. Here, we characterize the Av2 effector of the vascular wilt fungus Verticillium dahliae as a suppressor of the cry for help. Inspired by in silico antimicrobial activity prediction, we investigated the antimicrobial activity of Av2 in vitro. Furthermore, its role in V. dahliae virulence was assessed through microbiota sequencing of inoculated plants, microbial co-cultivation assays, and inoculations in a gnotobiotic plant cultivation system. We show that Av2 inhibits bacterial growth, and acts as a virulence factor during host colonization. Microbiota sequencing revealed involvement of Av2 in suppression of Pseudomonas spp. recruitment upon plant inoculation with V. dahliae, suggesting that Av2 suppresses the cry for help. We show that several Pseudomonas spp. are antagonistic to V. dahliae and sensitive to Av2 treatment. We conclude that V. dahliae secretes Av2 to suppress the cry for help by inhibiting the recruitment of antagonistic Pseudomonas spp. to pave the way for successful plant invasion.
|
|
Scooped by
?
June 11, 7:10 PM
|
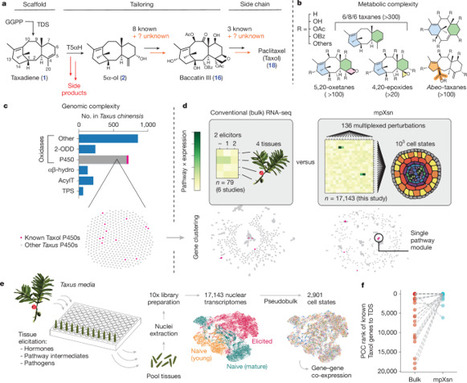
Plants make complex and potent therapeutic molecules, but sourcing these molecules from natural producers or through chemical synthesis is difficult, which limits their use in the clinic. A prominent example is the anti-cancer therapeutic paclitaxel (sold under the brand name Taxol), which is derived from yew trees (Taxus species). Identifying the full paclitaxel biosynthetic pathway would enable heterologous production of the drug, but this has yet to be achieved despite half a century of research. Within Taxus’ large, enzyme-rich genome, we suspected that the paclitaxel pathway would be difficult to resolve using conventional RNA-sequencing and co-expression analyses. Here, to improve the resolution of transcriptional analysis for pathway identification, we developed a strategy we term multiplexed perturbation × single nuclei (mpXsn) to transcriptionally profile cell states spanning tissues, cell types, developmental stages and elicitation conditions. Our data show that paclitaxel biosynthetic genes segregate into distinct expression modules that suggest consecutive subpathways. These modules resolved seven new genes, allowing a de novo 17-gene biosynthesis and isolation of baccatin III, the industrial precursor to Taxol, in Nicotiana benthamiana leaves, at levels comparable with the natural abundance in Taxus needles. Notably, we found that a nuclear transport factor 2 (NTF2)-like protein, FoTO1, is crucial for promoting the formation of the desired product during the first oxidation, resolving a long-standing bottleneck in paclitaxel pathway reconstitution. Together with a new β-phenylalanine-CoA ligase, the eight genes discovered here enable the de novo biosynthesis of 3’-N-debenzoyl-2’-deoxypaclitaxel. More broadly, we establish a generalizable approach to efficiently scale the power of co-expression analysis to match the complexity of large, uncharacterized genomes, facilitating the discovery of high-value gene sets. An approach that combines single-nucleus RNA sequencing and multiplexed perturbation identifies genes that enable the biosynthesis of direct precursors of the anti-cancer drug Taxol, whose current production involves a laborious extraction process from yew trees.
|
|
Scooped by
?
June 11, 6:37 PM
|
Phage therapy is emerging as a promising strategy against the growing threat of antimicrobial resistance, yet phage and bacteria are incredibly diverse and idiosyncratic in their interactions with one another. Clinical applications of phage therapy often rely on a process of manually screening collections of naturally occurring phages for activity against a specific clinical isolate of bacteria, a labor-intensive task that is not guaranteed to yield a phage with optimal activity against a particular isolate. Herein, we review recent advances in artificial intelligence (AI) approaches that are advancing the study of phage-host interactions in ways that might enable the design of more effective phage therapeutics. In light of concurrent advances in synthetic biology enabling rapid genetic manipulation of phages, we envision how these AI-derived insights could inform the genetic optimization of the next generation of synthetic phages.
|
|
Scooped by
?
June 11, 6:04 PM
|
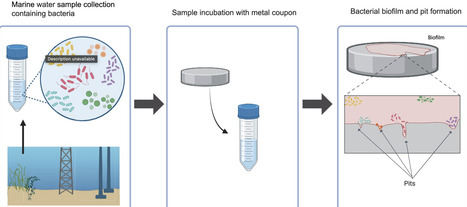
Steel corrosion is an extensive problem worldwide, substantially impacting marine infrastructures. In this study, the influence of steel on bacterial succession and corrosion was investigated by culturing marine water samples with and without steel coupons for 14 days. Compared to abiotic controls, oxygen levels were rapidly depleted in biotic cultures. Fe levels increased in controls compared to biotic cultures, potentially due to anoxic conditions and the incorporation of Fe in the biofilm. Proteobacteria dominated the initial cultures, but over 14 days the number of phylogenetic groups decreased overall in abundance. Taxons that increased in abundance included Clostridiaceae, Fusobacteriaceae, Flavobacteriaceae and Prolixibacteraceae, some members of which can utilize Fe. While initially in low abundance, Arcobacteraceae, Pseudoalteromonadaceae, Rhodobacteraceae and Rhizobiaceae numbers increased over time. Sites 1 and 2 cultures displayed localised deep pitting corrosion on coupon surfaces, consistent with microbial action, with an increase in Bacteroidetes, suggesting this phylum facilitates corrosion. In contrast, Site 3 cultures displayed uniform, superficial corrosion, with Clostridiaceae being the dominating family by Day 14, suggesting corrosion inhibition through biofilm formation. By identifying bacteria associated with corrosion, targeted approaches to corrosion reduction may be developed through identifying significant metabolic pathways by transcriptomics and the application of metabolic inhibitors.
|
|
Scooped by
?
June 11, 1:37 AM
|
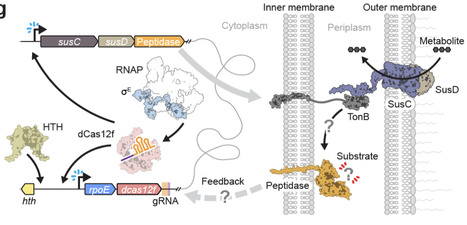
Bacterial transcription initiation is a tightly regulated process that canonically relies on sequence-specific promoter recognition by dedicated sigma (σ) factors, leading to functional DNA engagement by RNA polymerase (RNAP). Although the seven σ factors in E. coli have been extensively characterized, Bacteroidetes species encode dozens of specialized, extracytoplasmic function σ factors (σE) whose precise roles are unknown, pointing to additional layers of regulatory potential. Here we uncover an unprecedented mechanism of RNA-guided gene activation involving the coordinated action of σE factor in complex with nuclease-dead Cas12f (dCas12f). We screened a large set of genetically-linked dCas12f and σE homologs in E. coli using RIP-seq and ChIP-seq experiments, revealing systems that exhibited robust guide RNA enrichment and DNA target binding with a minimal 5ʹ-G target-adjacent motif (TAM). Recruitment of σE was dependent on dCas12f and guide RNA (gRNA), suggesting direct protein-protein interactions, and co-expression experiments demonstrated that the dCas12f-gRNA-σE ternary complex was competent for programmable recruitment of the RNAP holoenzyme. Remarkably, dCas12f-RNA-σE complexes drove potent gene expression in the absence of any requisite promoter motifs, with de novo transcription start sites defined exclusively by the relative distance from the dCas12f-mediated R-loop. Our findings highlight a new paradigm of RNA-guided transcription (RGT) that embodies natural features reminiscent of CRISPRa technology developed by humans.
|
|
Scooped by
?
June 11, 1:11 AM
|
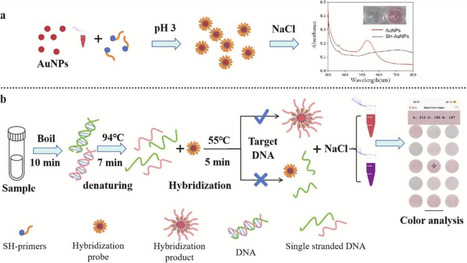
Total bacterial counts (TBC) and coliforms bacteria have been used as the index of microbial quality and fecal contamination for foods, the detection of them is involved in microbiological criteria of most countries to guarantee food safety. In this study, a gold nanoparticle (AuNPs) based colorimetric assay for rapid detection of TBC and coliform bacteria was developed by using the molecular hybridization approach. Thiol modified primers SH-27F and SH-lacZF were designed and conjugated with AuNPs, which was acted as both hybridization probe and signal probe, and salt induced aggregation of AuNPs was indicator of the hybridization and detection. Escherichia coli was studied as model strain in this study. The result showed that under the optimal conditions, the limit of detection for TBC and coliforms bacteria were 102 CFU/mL and 101 CFU/mL by visual detection, and 101 CFU/mL by UV–vis spectrum analysis, a smart phone application named ColorPicker was used for the quantitative analysis of the colorimetric assay. The color signal had good linear relationship of E. coli concentration in the range of 101–104 CFU/mL. The detection is rapid, simple and sensitive, without the needing of culture enrichment and precious equipment, has good application potential for the on-site detection of TBC and coliforms bacteria. In addition, the proposed strategy provides a direction for the design of highly sensitive colorimetric signal probes for other pathogenic microorganisms.
|
|
Scooped by
?
June 11, 1:02 AM
|
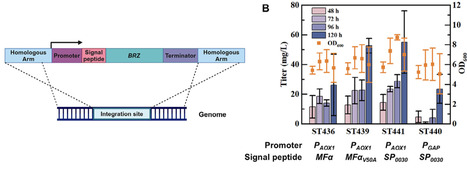
Excessive sugar consumption is associated with metabolic disorders such as obesity and diabetes, prompting the search for healthier alternatives. Sweet-tasting proteins offer high sweetness, safety, and low caloric content, making them promising sugar substitutes. However, their application is hindered by limited natural abundance and challenging extraction processes from plant sources. In this study, the microorganism-based sweet-tasting protein was explored. Brazzein was first verified as an expressible protein for production using the Komagataella phaffii expression system. Through the evaluation of promoters and signal peptides, the PAOX1→SP0030→PbBRZ→Tdas1 expression cassette was identified as optimal for brazzein production. Via the optimization of brazzein gene copy number and the modulation of protein translation, folding, and degradation processes, brazzein production was further improved to 113.24 and 166.54 mg/L, respectively. By performing fed-batch fermentation in a 5 L bioreactor, the engineered strain produced brazzein at a titer of 639.11 mg/L. This is the highest level for microbial brazzein production. These findings highlight the effectiveness of our strain optimization strategies and provide a foundation for the industrial application of sweet-tasting proteins.
|
|
Scooped by
?
June 11, 12:48 AM
|
Cupriavidus necator is a promising bacterium for producing polyhydroxybutyrate (PHB), a biodegradable bioplastic. However, the cell growth is restricted due to a lack of glucose transporters and low glucokinase activity. To address this limitation, we first developed comprehensive genetic toolkits to express sfGFP as a proof-of-concept in C. necator. A plasmid-driven T7 RNA polymerase (PDT7) system under the J23109 promoter achieved a 10-fold increase in fluorescence compared to strains without PDT7. However, PDT7 imposed a metabolic burden at higher expression levels and remained less effective than the constitutive Trc promoter, which consistently exhibited the highest transcriptional strength. Based on its robust and balanced performance, the Trc promoter was optimized to drive expression of galP (galactose permease) and glk (glucokinase) genes from Escherichia coli, (annotated as Tgg-H16), enabling enhanced glucose uptake, biomass and PHB biosynthesis. Further enhancement was achieved by supplementing a mixed carbon source, that is, 10 g/L glucose and 10 g/L fructose, which shortened the lag phase and supported higher PHB yields. Finally, a stepwise feeding strategy in fermentation boosted PHB production to 30.9 g/L. Rewiring carbon flux in C. necator through genetic circuit design demonstrates the feasibility of improving carbon uptake, while integration with tailored cultivation strategies enables upcycling sustainable PHB production.
|














genetic part