 Your new post is loading...

|
Scooped by
mhryu@live.com
Today, 3:28 PM
|
Argonaute proteins provide innate immunity in all domains of life through guide-dependent recognition and cleavage of invader nucleic acids. Many short prokaryotic Argonautes (pAgos) lack nuclease activity and are instead co-encoded with tentative nuclease effectors, but their activation mechanisms remain unknown. Here, we characterize SPARHA systems (short prokaryotic argonautes, HNH-associated), containing HNH nuclease effectors. RNA-guided target DNA recognition by pAgo induces formation of SPARHA filaments with a chain of double nuclease sites formed by HNH tetramers, leading to indiscriminate collateral degradation of DNA and protecting bacterial population from invaders. We show that the assembly of filaments proceeds via a universal activation pathway involving a cascade of target-induced conformational changes in SPARHA, conserved in other short pAgo systems containing various types of effectors. pAgos and associated effectors act as modular defense systems that translate recognition of specific DNA into immune response through assembly of supramolecular complexes, deleterious for invaders and potentially useful for biotechnology. Argonaute proteins degrade specific invader nucleic acids in eukaryotic and prokaryotic innate immunity. Here, Kanevskaya et al. describe a bacterial immune system in which RNA-guided recognition of invader DNA by Argonaute triggers formation of HNH nuclease filaments with collateral activity, protecting the bacterial population from invaders.
|
|
Scooped by
mhryu@live.com
Today, 2:56 PM
|
Natural CRISPR-Cas9 systems provides diverse properties for genome editing, yet finding compact variants remains a priority. In this study, we screened a panel of 11 CjCas9 orthologous using a GFP activation assay and identified seven active nucleases. Among these, Cj4Cas9 stood out as particularly noteworthy due to its compact genome size (985 amino acids) and unique PAM preference (5’-NNNGRY-3’). Cj4Cas9 demonstrates efficient disruption of the Tyr gene in mouse zygotes, resulting in an albino phenotype. Furthermore, when delivered via AAV8, Cj4Cas9 achieves efficient genome editing of the Pcsk9 gene in mouse liver, leading to reduced serum cholesterol and LDL-C levels. Seeking to further expand its utility, we engineered Cj4Cas9 for higher activity by introducing L58Y/D900K mutations, resulting in a variant termed enCj4Cas9. This variant exhibits a two-fold increase in nuclease activity compared to the wild-type Cj4Cas9 and recognizes a simplified N3GG PAM, considerably expanding its targeting scope. These findings establish Cj4Cas9 and its engineered variants for fundamental research and therapeutic applications. A compact Cas9 ortholog, Cj4Cas9, enables efficient genome editing in vivo compatibility.
|
|
Scooped by
mhryu@live.com
Today, 2:35 PM
|
With the rise in antimicrobial resistance, understanding the virulence factors utilized by pathogenic E. coli is essential for the development of alternative therapeutics. While previous work has shown that disruption of the E. coli rhomboid protease gene glpG leads to defects in bacterial colonization, here we provide mechanistic insight into the loss of fitness. We show GlpG is essential for the assembly of type 1 pili, a virulence factor required for the colonization of eukaryotic cells. Since pili are critical for biofilm formation and bacterial persistence, the absence of GlpG proteolytic activity reduces the production of biofilm. Working towards new potential antimicrobial targets for treating infections, we show that biofilm formation is hampered by GlpG inhibition. Our data demonstrates that GlpG plays a key role in protein quality control of type 1 pili and alters the paradigm for GlpG proteolysis, previously implicated in the cleavage of only membrane embedded substrates. The disruption of the rhomboid protease gene glpG in E. coli leads to defects in bacterial colonization. Here, the authors show that GlpG is essential for the assembly of type 1 pili, a virulence factor required for colonization of bacteria in eukaryotic cells.
|
|
Scooped by
mhryu@live.com
Today, 2:22 PM
|
Droplet microfluidics is a core technology that powers high-throughput single cell sequencing. However, the current generation of single-cell microfluidics faces notable limitations, including cell aggregation, suboptimal on-chip reactions that compromise experimental outcomes and elevate background noise, as well as a dependence on costly commercial barcode beads. To address these challenges, we present MusTer, an integrated next-generation platform with multi-parametric singlet droplet sorting and triple-droplet merging capability. MusTer's multi-parametric singlet sorting module enables in-line droplet analysis of intrinsic fluorescence peak amplitude, width and interval from single-nucleus- (singlet) or multiple-nuclei (multiplet)-encapsulating droplets, subsequently allowing an effective separation of the singlet droplets from multiplet droplets and empty droplets. MusTer's triple-droplet merging module enables precise multi-step reactions, with each step performed under its own optimal conditions, thereby significantly enhancing experimental flexibility and efficiency. We validated MusTer's performance by performing single-cell ATAC-seq on maize leaves. The results demonstrate that MusTer significantly reduces the doublet rate, enhances the signal-to-noise ratio, and yields improved cell clustering compared with traditional methods. These results validate MusTer's capability to overcome key limitations in droplet-based single-cell analysis, effectively enhancing data quality and reliability, and also paves the way for its use in other challenging sample types and multi-step single-cell assays.
|
|
Scooped by
mhryu@live.com
Today, 1:16 PM
|
Temperature offers a simple yet powerful signal to program cellular behavior. Here, we engineered and characterized a set of temperature-dependent genetic circuits that integrate RNA thermometers with site-specific DNA recombinases to achieve precise, irreversible control of gene expression. Using the serine recombinase Bxb1 placed under the control of the Salmonella FourU RNA thermometer, we demonstrate how promoter strength critically defines thermal sensitivity: weak promoters’ activity clears ON/OFF transitions, while strong promoters lead to continuous, quasi-temperature-independent recombination. Furthermore, temperature pulse duration and growth phase of cell culture were found to modulate recombination efficiency, providing additional layers of control. We illustrate the potential of this framework through proof-of-concept applications, including (i) the generation of spatial expression patterns on 2D surfaces via localized heating, (ii) a paper-based device capable of recording temperature gradients as stable genetic outputs, and (iii) a temperature-triggered lysis system for controlled cellular release. Together, these results establish temperature-regulated recombinase circuits as versatile and robust tools for programmable, spatially resolved, and irreversible control of gene expression, paving the way for new applications in synthetic biology, biosensing, and bioproduction.
|
|
Scooped by
mhryu@live.com
Today, 11:43 AM
|
Gene doping is an increasing challenge in sports, demanding highly sensitive and specific detection tools beyond the limitations of the current amplification-dependent methods. Here, an innovative amplification-free CRISPR-Cas12a assay integrated with osmotically tunable double emulsion (DE) droplets is reported for rapid and ultrasensitive gene doping detection. Target DNA and CRISPR/Cas12a complexes are encapsulated within DE droplets, where osmotic shrinkage rapidly concentrates the reaction components, thereby enhancing the fluorescent signal intensity without nucleic acid amplification. This platform enables the detection of the human erythropoietin (hEPO) gene at unprecedented attomolar levels within 30 min, achieving a 25-fold improvement in sensitivity compared with that of nonshrinkable formats. Notably, the assay demonstrated a robust and specific performance in complex serum samples with minimal matrix interference. This novel approach offers a rapid, reliable, and inherently contamination-free solution for gene doping surveillance with broad potential for versatile amplification-free nucleic acid diagnostics.
|
|
Scooped by
mhryu@live.com
December 29, 11:34 PM
|
Biomaterials are gaining attention as sustainable alternatives in the fashion industry. In a recent Trends in Biotechnology article, Zhou et al. optimized a one-pot strategy for the growth of colored bacterial cellulose, enabling more sustainable production of textile materials. Here, we contextualize this development alongside other innovations and roadblocks in sustainable fashion.
|
|
Scooped by
mhryu@live.com
December 29, 10:46 PM
|
Design of experiments (DOE) principles are increasingly applied to biological assays, yet it remains unclear whether their foundational assumption, orthogonal decomposition, holds in nonlinear biological systems. We addressed this question using Perturb-seq as a case study. By benchmarking a design commonly used in Perturb-seq and related experiments against the orthogonal Plackett-Burman (PB) design via simulations, we uncovered a counter-intuitive phenomenon: while orthogonal designs generally excel, the multicollinearity inherent to the common design is functionally advantageous in systems with significant signal amplification. This challenges the blind application of DOE to biology. Based on these findings, we developed the PB suitability index (PBSI), a simple, parameter-free metric that predicts the optimal design solely from network structure. Our work not only provides practical guidelines for Perturb-seq but also establishes a "biology-oriented DOE" framework, bridging the gap between statistical rigor and biological complexity.
|
|
Scooped by
mhryu@live.com
December 29, 10:39 PM
|
Microorganisms underpin global biogeochemical cycles and represent a vast, underexplored reservoir for sustainable biotechnology and human health. Their diversity, arising from the interplay of numerous genes within holistic genomic contexts, dictates complex ecophysiological traits across varied environments. To bridge the gap between complex genomic sequences and associated biological functions and phenotypic outcomes, we present MicroGenomer, a foundation model for transferable microbial genome representations enabling multi-scale genomic understanding and ecophysiological trait prediction. MicroGenomer leverages a hierarchical training strategy comprising pre-training on large-scale genomic sequences (234.5 billion base pairs), domain-specific mid-training using the GTDB-curated marker gene set, and task-specific post-training. Despite a streamlined architecture of 470 million parameters, MicroGenomer achieves performance on key tasks competitive with models nearly 85 times its size. Advancing beyond established gene-scale encoders, MicroGenomer generates robust embeddings at the genome scale for downstream modeling. Extensive evaluations demonstrate that MicroGenomer effectively captures phylogenetic structures in species space, excelling in gene- and genome-scale understanding as well as ecophysiological trait prediction. The practical utility of these capabilities is further demonstrated by targeted wet-lab validation on newly isolated strains, indicating that MicroGenomer's predictions provide reliable guidance for biological experiments. Collectively, MicroGenomer offers a high-performance, resource-efficient framework that transforms raw sequence data into actionable biological insights, providing a powerful foundation for microbiome research and biotechnology.
|
|
Scooped by
mhryu@live.com
December 29, 3:03 PM
|
Antimicrobial peptides (AMP) are crucial in addressing the global crisis of bacterial resistance. However, there are still significant limitations in existing methods on de novo AMPs design, especially in designing AMPs with desirable physicochemical properties for specific bacterial pathogens. In this study, we propose a novel generative framework for designing pathogen-targeted antimicrobial peptides with programmable physicochemical properties. More specifically, a conditional Variational Autoencoder is first pretrained for generating AMPs with editable physicochemical properties. We then develop a conditional diffusion model to learn hidden representations of AMPs for targeting pathogens of interest, and construct corresponding MIC predictors for specific bacterial strains. Through comprehensive simulation experiments, we demonstrate that the proposed framework outperforms most existing models in terms of antimicrobial efficacy against specific bacterial targets. Moreover, through systematic screening and analysis, we have identified two star AMPs for each of the two target bacterial species (i.e., E. coli or S. aureus), both of which exhibit excellent performance in antibacterial activity, hemolytic properties, toxicity profiles, etc. Overall, this study provides the key technological support for developing next-generation intelligent platforms for antimicrobial agents design.
|
|
Scooped by
mhryu@live.com
December 29, 2:55 PM
|
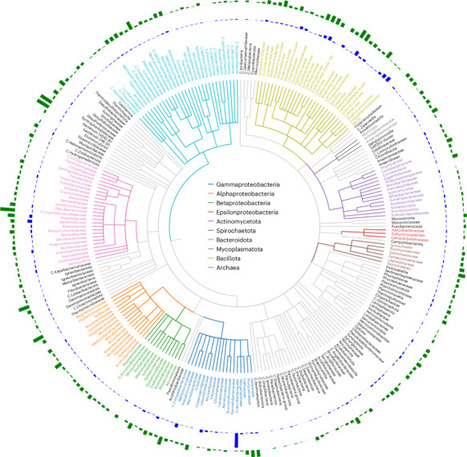
Phages are typically classified as temperate, integrating into host genomes, or lytic, replicating and killing bacteria; for this reason, lytic phages are not expected in bacterial genome sequences. Here we analyse 3.6 million bacterial genome assemblies from 1,226 species and find 119,510 lytic phage genomes, which we term bacterial assembly-associated phage sequences. This represents a ~5-fold increase in the number of phages with associated hosts and raises questions about fundamental aspects of phage biology. Our analyses of bacterial assembly-associated phage sequences revealed previously undescribed phage clusters, including clusters distantly related to Salmonella Goslarviruses in Escherichia coli and Shigella, while also substantially expanding known genera such as Seoulvirus (from 16 to >300 members). Close relatives of lytic phages used therapeutically were also detected, suggesting clinical isolate sequencing unknowingly archives potential phage candidates. The discovery of complete, lytic phage genomes within bacterial assemblies challenges assumptions about the nature of the lytic lifestyle and reveals an untapped reservoir of phages. Diverse genomes of lytic phages are found in bacterial assemblies, challenging assumptions about the nature of the lytic lifestyle.
|
|
Scooped by
mhryu@live.com
December 29, 2:48 PM
|
Untargeted, also known as metagenomic, nanopore sequencing is a powerful tool for virus genomic surveillance, particularly in resource-limited settings and when paired with the portability of the MinION device (Oxford Nanopore Technologies, ONT). However, a major bottleneck for global access is the absence of a user-friendly software capable of efficiently analyzing untargeted nanopore sequencing data to generate high-quality consensus genomes. We share ViMOP, a pipeline built on our long-term experience in nanopore field sequencing. The pipeline emphasizes field user-friendliness, flexibility and versatility to analyze reads generated directly from human clinical samples. The software assembles de novo contigs, matches contigs to known viral references and uses them to assemble consensus genomes. Executed with a single Nextflow command or via the EPI2ME Desktop interface (ONT), results are summarized in an HTML report. ViMOP, through its user-centered design, lowers the barrier to high-quality virus genome reconstruction and advances capacity for genomic surveillance.
|
|
Scooped by
mhryu@live.com
December 29, 2:30 PM
|
Sporulation is a strategy employed by many bacteria to survive harsh environmental conditions. The genus Paenibacillus includes spore-forming species notorious for spoiling pasteurized dairy products and for causing American foulbrood in honeybee larvae, leading to colony collapse. Human pathogens within Paenibacillus are also a growing threat, causing fatal opportunistic infections. Here, we present a comprehensive survey of sporulation genes across 1,460 high-quality Paenibacillus genomes. We find that all members of the sporulation-initiating phosphorelay are well conserved, but that the Spo0B phosphotransferase contains a predicted transmembrane domain. We confirm that this domain localizes Spo0B to the cell membrane and therefore refer to this Spo0B variant as Spo0B-TM. Spo0B-TM is present in 92% of surveyed Paenibacillus genomes. Consistent with its high level of conservation, we find that the transmembrane domain is important for detecting its interaction with its phosphorelay partners Spo0A and Spo0F. Moreover, we find that Spo0B exhibits low sequence identity across Bacillota when compared with other members of the phosphorelay. Altogether, this work highlights the potential for diversity even within the highly conserved phosphorelay that initiates sporulation in Bacillota.
|
|
|
Scooped by
mhryu@live.com
Today, 3:09 PM
|
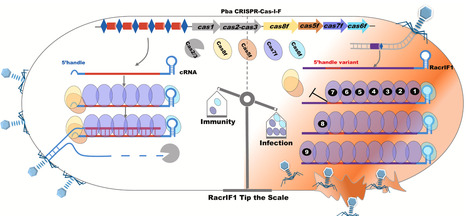
RNA-based anti-CRISPRs (Racrs) interfere with the type I-F CRISPR-Cas system by mimicking the repeats found in CRISPR arrays. Here, we determined the cryo-electron microscopy (cryo-EM) structures of the type I-F crRNA-guided surveillance complex (Csy complex) from Pectobacterium atrosepticum and three RacrIF1-induced aberrant subcomplexes. Additionally, we observed that Cas7f proteins could bind to non-specific nucleic acids, forming right-handed superhelical filaments composed of different Cas7 copies. Mechanistically, RacrIF1 lacks the specific S-conformation observed in the corresponding position of the 5′ handle in canonical CRISPR complexes, and it instead adopts a periodic “5 + 1” pattern. This conformation creates severe steric hindrance for Cas5f–Cas8f heterodimer and undermines their binding. Furthermore, Cas7f nonspecifically binds nucleic acids and can form infinite superhelical filaments along Racrs molecules. This oligomerization sequesters Cas6f and Cas7f from binding, therefore blocking the formation of functional CRISPR-Cas effector complexes and ultimately blocking antiviral immunity. Our study provides a structural basis underlying Racrs-mediated CRISPRs inhibition.
|
|
Scooped by
mhryu@live.com
Today, 2:41 PM
|
Nitrogen availability is a key determinant of plant growth and development. Here, we investigate how different N sources shape Arabidopsis thaliana root system architecture, metabolism and hormone dynamics. L-glutamine (L-GLN) significantly enhances root biomass compared to nitrate (KNO3) without compromising shoot growth. This effect emerges after 2 weeks and is independent of L-GLN's role as a carbon or ammonium source or of potential L-GLN-induced pH changes due to ammonium release, indicating a specific function of L-GLN as a N source and signaling molecule. A reverse genetic screen identified AMINO ACID PERMEASE 1 (AAP1)-mediated uptake and GLUTAMINE SYNTHETASE (GS)-dependent assimilation as essential for L-GLN-induced root biomass. In contrast, the N-sensing regulators NITRATE TRANSPORTER 1.1 (NRT1.1) and AMMONIUM TRANSPORTER (AMT) family members contribute to the differential root responses between KNO3 and L-GLN. Metabolic profiling revealed distinct amino acid signatures under these N sources, irrespective of genotype. Hormonal analyses showed that L-GLN modulates auxin homeostasis, with auxin supplementation restoring primary root growth and lateral root symmetry under L-GLN conditions. L-GLN also reconfigures cytokinin balance by elevating cZ while reducing tZ, collectively shaping root system architecture through hormone-dependent regulation. Together, these findings establish L-GLN as an integrator of N metabolism and hormone signaling in root development, highlighting its signaling capacity beyond nutrient supply and offering new perspectives for improving N use efficiency.
|
|
Scooped by
mhryu@live.com
Today, 2:26 PM
|
3'-5' translation (termed "backward translation") mediated by eukaryotic circular RNAs has been reported recently. Whether linear mRNA can produce proteins through backward translation remained elusive. Here, we demonstrate that linear mRNAs can mediate backward translation in vitro. Backward translation was detected in human cells transfected with expression vectors used to produce linear mRNAs. Importantly, synthetic linear mRNAs could produce backward translation signal in multiple in vitro translation systems, with a level comparable to that of conventional 5' to 3' forward translation. Furthermore, we demonstrated that a single translation initiating sequence (TIS) was able to drive backward and forward translation in an in vitro experimental setup, with the efficiency of backward translation found to be significant lower than that of forward translation. In summary, this study further reinforces the notion of translation flexibility, demonstrates a broader applicability of backward translation and heralds a new strategy in synthetic biology.
|
|
Scooped by
mhryu@live.com
Today, 2:10 PM
|
MicroRNAs (miRNAs) are key posttranscriptional regulators of gene expression that influence cancer initiation, progression, and therapeutic response. While most studies have focused on endogenous miRNAs, emerging evidence has highlighted the role of plant-derived miRNAs as exogenous dietary regulators capable of cross-kingdom gene modulation. This review summarises current knowledge regarding plant-derived miRNAs and their ability to regulate human cancer-related genes. Experimental findings indicate that plant miRNAs can withstand gastrointestinal digestion, enter the circulation, and regulate the expression of oncogenes, tumor suppressors, long noncoding RNAs, and immune checkpoint molecules via canonical RNA-induced silencing mechanisms. Specific examples include miR-156a, miR-159a-3p, miR-166a, miR-167e-5p, miR-171, miR-395e, miR-2911, miR-4995 and miR-5754, which exhibit anticancer activities across various cancer types and modulate key signalling pathways in mammalian cells, highlighting their potential as cross-kingdom regulators with therapeutic relevance. In addition to these characterised miRNAs, certain plant groups, which are rich in bioactive compounds, remain unexplored as sources of functional miRNAs, representing a promising avenue for future research. Collectively, these studies underscore the ability of plant-derived miRNAs to modulate mammalian gene expression and suggest their potential as diet-based or synthetic therapeutic agents. Further investigations into their bioavailability, target specificity, and functional relevance could inform innovative strategies for cancer prevention, integrating nutritional, molecular biological, and therapeutic approaches.
|
|
Scooped by
mhryu@live.com
Today, 11:57 AM
|
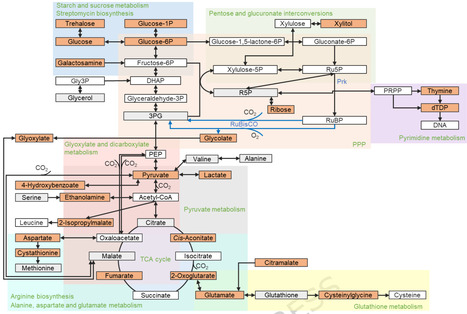
Rising atmospheric CO₂ levels and their impact on climate change have intensified the need for innovative carbon capture and fixation strategies. The Calvin-Benson-Bassham (CBB) cycle, a central metabolic pathway in all photoautotrophic organisms and many autotrophic bacteria, plays a pivotal role in global carbon assimilation but is limited by the low catalytic efficiency of Rubisco. Here, we engineered a complete, functional CBB cycle in Escherichia coli, by heterologously expressing up to 13 genes encoding phosphoribulokinase, α-carboxysomes, and inorganic carbon pumps. This bioengineering approach allowed E. coli to utilize atmospheric CO2 and led to increased levels of sugars such as ribose (4.94-fold) and xylitol (8.94-fold). Detailed metabolomic profiling of central carbon metabolism using gas chromatography-mass spectrometry (GC-MS) demonstrated that installation of the CBB cycle has a notable impact on the metabolic landscape of E. coli, resulting in substantial alterations in central carbon and amino acid metabolism. These findings deepen our understanding of the natural biological carbon-fixation pathway and its engineering in heterotrophic hosts. Furthermore, this work provides a versatile platform for evaluating and selecting efficient carbon-fixation modules, as well as assessing metabolic bottlenecks in engineered systems. These advances offer practical guidance for rational metabolic engineering in diverse organisms for biotechnological applications, including carbon sequestration, sustainable bioproduction, and crop improvement.
|
|
Scooped by
mhryu@live.com
Today, 11:30 AM
|
Escherichia coli is a promising host for terpenoid production, yet kinetic models tailored for such strains are limited—hindering effective bioprocess optimization and control. To address this, we developed a kinetic model of E. coli engineered to produce viridiflorol, aiming to capture the dynamics of key bioprocess variables under both uninduced and induced conditions. To account for growth and metabolism of cells, a simplified central metabolic pathway of E. coli was considered by including acetyl-CoA as the intracellular metabolite due to its role as a common precursor for viridiflorol, acetate, and the TCA cycle. Initial modeling efforts based solely on metabolism failed to reproduce experimental trends, prompting the inclusion of cellular stress caused by IPTG-induced heterologous pathway activation. However, this adjustment alone could not fully explain the observed trends, leading us to hypothesize the emergence of two distinct subpopulations post-induction: (1) stressed, slow-growing producers and (2) dormant non-producers that eventually outgrow producers. After iterative refinements, the model successfully replicated experimental trends with R2 value for glucose, cells, acetate, dissolved oxygen, and viridiflorol being 0.98, 0.88, 0.86, 0.70, and 0.94 respectively. Finally, model simulations were performed for insights which suggested that lowering IPTG concentration can improve viridiflorol production by delaying the emergence of the non-producer population. Guided by the simulations, an optimal IPTG concentration (9.375 µM) for a batch condition was identified, resulting in a titre of 0.14 g/L of viridiflorol. In contrast, a non-optimal IPTG concentration (150 µM) yielded a titre of only 0.03 g/L. Furthermore, experimental validation of the subpopulation hypothesis showed the coexistence of producer and non-producer populations. Taken together, the model captured the bioprocess variable trends, predicted an optimal IPTG concentration for a batch process, and provided insights into the existence of two subpopulations.
|
|
Scooped by
mhryu@live.com
December 29, 10:55 PM
|
Solanoeclepin A (SolA) is a triterpenoid exuded from the roots of Solanaceae plants, originally identified as hatching stimulant for plant parasitic cyst nematodes. Assuming that evolution would have selected against the production of such a fitness lowering molecule, we postulated that SolA must serve another, beneficial, role for the plant. In this study, we demonstrate that nitrogen (N) deficiency strongly increases the SolA concentration in tomato root exudate. Moreover, SolA is produced only under non-sterile conditions, indicating that soil microbiota are involved in its production. Time-resolved RNAseq analysis revealed several candidate genes for SolA biosynthesis, which were all upregulated under N deficiency. Transient silencing of two SolA biosynthetic genes (e.g., CYP749A19 and CYP749A20) significantly reduced SolA production. Microbiome analysis on the rhizosphere of these plants demonstrated that the recruitment of beneficial Massilia spp. was inhibited in the transiently silenced plants. Isolation of a Massilia strain, identified as Massilia cellulosiltytica, allowed us to show that it has growth-promoting activity under N deficiency, likely via indole-3-acetic acid production and enhanced N acquisition. Root exudate of N-starved tomato displayed strong chemotactic activity towards this strain. Together, these findings demonstrate that SolA production under N deficiency relies on the interaction between tomato and soil microbiota that convert a plant-produced precursor that is a recruitment signal for beneficial, growth-promoting microbes to SolA, which was hijacked by cyst nematodes as a reliable host presence cue. This dual functionality resembles the microbial transformation of primary into secondary bile acids, important signalling molecules, in the gut of animals, and suggests convergent evolution in host microbe co-metabolism. Overall, our study positions SolA as a multifunctional signaling molecule in rhizosphere interactions, with potential application for enhancing crop resilience and sustainable pest management.
|
|
Scooped by
mhryu@live.com
December 29, 10:43 PM
|
The identification of antifungal proteins (AFPs) is crucial for understanding microbial interactions and facilitating the discovery of antifungals on a large scale from genomic and metagenomic data. Most existing computational tools are developed for predicting antifungal peptides rather than proteins. To address this limitation, we developed AntiFP2. A manually curated dataset of experimentally validated AFPs was used to train and develop prediction models using cross-validation techniques. The ensemble strategy, which combined ESM2, BLAST, and MERCI, achieved the best results across independent validation. Beyond prediction, we implemented a complete pipeline for genome- and metagenome-wide AFP screening. AntiFP2 is freely available as a web server, standalone package, and Docker container, enabling scalable and reproducible antifungal protein discovery.
|
|
Scooped by
mhryu@live.com
December 29, 3:07 PM
|
Heterotrophic nanoflagellates are the chief agents of bacterivory in the aquatic microbial loop but remain underrepresented in culture collections and in genomic databases. We isolated and characterized a representative of the previously uncultured freshwater Cryptomonad Group 1 (CRY1a) lineage using a genome-streamlined, ultra-small and abundant microbe Planktophila versatilis as a prey and Catalyzed Reporter Deposition-Fluorescence in situ Hybridization (CARD-FISH) probe–based screening. This isolate, Tyrannomonas regina, is one of the most dominant ubiquitous heterotrophic cryptomonads in freshwaters. It is a small heterotrophic nanoflagellate (ca. 3–5 μm) and has the smallest genome of any cryptomonad sequenced thus far. The compact genome (ca. 69 Mb) revealed no traces of a photosynthetic lifestyle, consistent with its phylogenomic placement as a sister clade to cryptophytes that are characterized by the acquisition of a red-algal symbiont. Moreover, in comparison to its photosynthetic counterparts, its genome presents substantially lower repeat content and endogenous viral elements. Genomes of two giant viruses, Tyrannovirus reginensis GV1 and GV2, were also recovered from the same culture and represent a viral genus that has been described so far solely by metagenome-recovered genomes. Collectively, these findings provide insights into genomic ancestry and evolution, widespread ecological impact, and interactions of an elusive protist lineage and illustrate the advantages of culture-centric approaches towards unfolding complex tapestries of life in the microbial world.
|
|
Scooped by
mhryu@live.com
December 29, 3:01 PM
|
Coordinating multiple liquid handling robots is a complex logistical task when designing biological experiments. Protocol designers must consider the capabilities and constraints of each robot to distribute work optimally across multiple instruments. We developed an optimization framework that finds optimal liquid handling solutions that leverage an arbitrary number of robots. Our algorithm, called Pourfecto, abstracts the capabilities of each robot and their labware, allowing us to plan and schedule a wide range of biological experiments using commercial instruments and custom-built hardware. Pourfecto can optimize multiple objectives (minimum transfers, fewest reagents, fewest labware swaps) and scales to experiments with hundreds of thousands of liquid transfers.
|
|
Scooped by
mhryu@live.com
December 29, 2:51 PM
|
Bacteria have a variety of mechanisms for limiting predation by phages. SpbK is a Toll/interleukin-1 receptor (TIR) domain-containing antiphage defence protein from Bacillus subtilis that provides protection against the temperate phage SPβ via abortive infection. Here we structurally characterise SpbK and its interaction with the SPβ protein YonE. We demonstrate that SpbK is an NADase that produces both ADP-ribose (ADPR) and canonical cyclic ADPR with a N1-glycosidic bond (cADPR, also referred to as N1-cADPR). Combining cryo-EM, in silico predictions, site-directed mutagenesis, and phage infection assays, we show that formation of two-stranded head-to-tail assemblies of SpbK TIR domains is required for both NADase activity and antiphage defence. We also demonstrate that YonE is a dodecameric portal protein that activates the NADase function of SpbK by facilitating TIR domain clustering. Collectively, our results provide insight into how bacterial TIR NADases recognise phage infection. SpbK protects Bacillus subtilis from phage infection by depleting NAD⁺. In this study, the authors uncover the molecular mechanisms underlying SpbK’s self association-dependent NADase activity and its activation by the SPβ phage portal protein YonE.
|
|
Scooped by
mhryu@live.com
December 29, 2:41 PM
|
Microbial consortia show promise for bioremediation of environmental pollution, but performance optimization and risk assessment remain challenging due to unculturable species and limitations of traditional biochemical and sequencing tools. This study demonstrates how a multi-omics approach can provide deeper insight into the performance and risks of using a model aerobic ammonia-oxidizing consortium under conditions representative of wastewater treatment. Long-read DNA sequencing recovered several high-quality genomes, revealing dominance by an unclassified Nitrosospira species with expected ammonia oxidation capabilities. Lower-abundance taxa with nitrogen cycling potential were also detected, though species-level identification was limited by poor taxonomic database representation. Multi-omics and nitrogen analyses showed shifts in community composition and nitrogen cycling activity when the consortium was grown along a redox gradient typical of wastewater. All cultures accumulated ammonia over 4 weeks, with only aerobic cultures reducing ammonia levels thereafter. The dominant Nitrosospira population declined in abundance and activity in aerobic cultures while shifting toward nitrogen reduction under anoxic conditions. This metabolic shift would not have been detected using amplicon sequencing alone. Multi-omics also supported risk assessment through detection of waterborne pathogens from the Legionella genus and other lineages harboring virulence genes resembling those from known pathogens. This study highlights the value of multi-omics for optimizing microbial consortia and assessing biosafety risks but also underscores challenges related to effective data analyses and the feasibility of risk assessment under realistic conditions. Addressing these challenges will be essential to support the broader adoption of multi-omics strategies by stakeholders working with microbial consortia across diverse environmental applications.
|






























kortemme, synthetic protein design