 Your new post is loading...

|
Scooped by
mhryu@live.com
Today, 1:17 PM
|
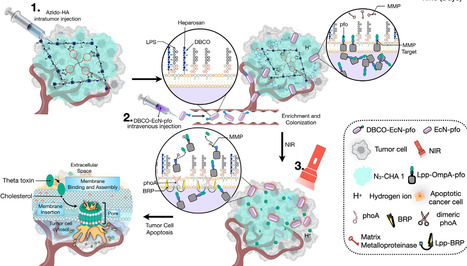
Rapid advances in synthetic biology are driving the development of microbes as therapeutic agents. While the immunosuppressive tumor microenvironment creates a favorable niche for the systematic delivery of bacteria and therapeutic payloads, these can be harmful if released into healthy tissues. To address this limitation, we designed a spatiotemporal targeting system for engineered Escherichia coli Nissle 1917, controlled by azide-modified hyaluronic acid hydrogel and near-infrared radiation induction. Using a temperature-driven genetic status switch, the system produced durable therapeutic output and promoted the therapeutic activity in solid tumors. The combination of azide-modified hyaluronic acid hydrogel and temperature-sensitive, engineered Escherichia coli Nissle 1917 provided spatiotemporal targeting of solid tumors, not only showing significant therapeutic effects on primary solid tumors, but also inhibiting the metastasis and recurrence of cancer cells by enhancing tumor-infiltrating lymphocytes. This system has potential for clinical application.
|
|
Scooped by
mhryu@live.com
Today, 12:55 PM
|
Understanding how plants perceive their environment is fundamental to advancing agricultural productivity and sustainability. Many biological small molecules, including those involved in microbial recognition, act rapidly at the plant cell surface, but the absence of tools to visualize these dynamics has limited our ability to dissect plant–microbe communication. To address this gap, we sought to create a genetically encoded biosensor that couples ligand-induced protein dimerization with the production of a fluorescent reporter. Inteins are peptide regions that excise themselves from precursor proteins and ligate the flanking chains (exteins). When each half of a split intein is fused to one of two dimerizing proteins, ligand binding brings them into proximity, inducing intein splicing and ligation of flanking extein sequences. We split the yeast vacuolar ATPase subunit 1 (VMA1) intein, creating a protein biosensor that produces eGFP upon protein dimerization after ligand binding. Specifically, eGFP halves (i.e., non-functional N- and C-terminal GFP fragments) were fused to the intein halves, resulting in two fusion proteins: N-terminal GFP::N-terminal intein and C-terminal intein::C-terminal GFP. To adapt our biosensor framework for chitin detection, we leveraged this chitin-sensing pathway by attaching two biosensor halves (N-terminal eGFP::N-terminal intein and C-terminal intein::C-terminal eGFP) to LYK5 and CERK1, respectively, creating a chitin sensor. If chitin detection occurs, eGFP will reassemble directly attached to LYK5, which is anchored in the plant cell membrane.
|
|
Scooped by
mhryu@live.com
Today, 11:43 AM
|
Natural healthy food colorants are increasingly demanded to replace synthetic ones which are linked to public health concerns. Although nature provides a rich palette of colors, producing food colorants from food crops or specific agricultural products can exacerbate food security issues and environmental challenges, such as deforestation driven by the expansion of farmland. Synthetic biology offers promising solutions to address the supply limitations of natural food colorants through developing color-enriched plant varieties and particularly, high-yield microbial cell factories for production through precision fermentation. For the efficient translation of microbial fermentation solutions, it is important to integrate interdisciplinary synthetic/engineering biology research platforms, ranging from AI-assisted biological design to streamlined strain engineering workflows, and systems biology facilities, to address production challenges for scaling up. Bridging the lab-to-market gap through innovative economics and legality mechanism can accelerates the industrialisation of sustainable natural food colourants.
|
|
Scooped by
mhryu@live.com
December 31, 2025 10:21 PM
|
The MarR-family regulator MhqR of Staphylococcus aureus (SaMhqR) was previously characterized as a quinone-sensing repressor of the mhqRED operon. Here, we solved the crystal structures of apo-SaMhqR and the 2-methylbenzoquinone (MBQ)-bound SaMhqR complex. AlphaFold3 modeling was used to predict the structure of SaMhqR in complex with its operator DNA. In the DNA-bound SaMhqR state, S65 and S66 of an allosteric α3–α4 loop adopted a helically wound conformation to elongate helix α4 for optimal DNA binding. Key residues for MBQ interaction were identified as F11, F39, E43, and H111, forming the MBQ-binding pocket. MBQ binding prevented the formation of the extended helix α4 in the allosteric loop, leading to steric clashes with the DNA. Molecular dynamics (MD) simulations revealed an increased intrinsic dynamics within the allosteric loop and the β1/β2-wing regions after MBQ binding to prevent DNA binding. Using mutational analyses, we validated that F11, F39, and H111 are required for quinone sensing in vivo, whereas S65 and S66 of the allosteric loop and D88, K89, V91, and Y92 of the β1/β2-wing are essential for DNA binding in vitro and in vivo. In conclusion, our structure-guided modeling and mutational analyses identified a quinone-binding pocket in SaMhqR and the mechanism of SaMhqR inactivation, which involves local structural rearrangements of an allosteric loop and high intrinsic dynamics to prevent DNA interactions. Our results provide novel insights into the redox mechanism of the conserved SaMhqR repressor, which functions as an important determinant of quinone and antimicrobial resistance in S. aureus.
|
|
Scooped by
mhryu@live.com
December 31, 2025 10:06 PM
|
Transcriptional control arises from the specific recognition of promoter DNA by transcription factors (TFs), forming the basis of cellular information processing and gene regulation. In synthetic biology, TF-promoter interactions are assembled into gene circuits to program cellular behaviors. To ensure reliable circuit performance, most synthetic gene circuits rely on well-characterized and orthogonal regulatory parts. This reliance minimizes crosstalk but constrains circuit complexity and information integration. Creating hybrid TFs that combine or interpolate promoter specificities could therefore expand the design space of synthetic regulatory systems. However, it remains unclear whether hybrid functions can be created by mixing amino acid sequences, and how such functional integration could be achieved in a principled manner. Here we show that a variational autoencoder (VAE) trained on LuxR-family DNA-binding domains can generate transcription factors with hybrid and partially novel promoter recognition properties. By sampling intermediate regions of the VAE-learned latent space, we designed hybrid TFs that activate both the lux and las promoters. High-throughput sort-seq assays together with individual in vivo assays revealed that a subset of functional variants exhibited dual-responsive behavior while maintaining sequence-selective DNA recognition. Together, these results provide a data-driven strategy for exploring functional intermediate sequences between closely related proteins.
|
|
Scooped by
mhryu@live.com
December 31, 2025 9:48 PM
|
Genetic modules are often designed and implemented with inspiration from engineering disciplines. Although this approach can be successful because of the similarities underpinning physical and biochemical systems, it neglects a key factor that affects the performance of living organisms: evolution. Thus, it is crucial to incorporate the impact of inevitable mutations into the design and analysis of genetic modules. Combining computational modeling and in vivo mutagenesis experiments in Escherichia coli, we characterize how the interplay of gene dosage via plasmid copy number (PCN) and regulatory architecture affect the phenotypic mutation rate. For example, while greater PCN facilitates the emergence of gain-of-function mutations, it instead curbs the spread of loss-of-function mutations. We further reveal that mutations in the coding region are often masked at the phenotypic level, unlike those occurring in the regulatory region which become more prominent as PCN increases, both when the regulator is expressed constitutively and when it is self-repressed. Together, our results shed light on evolutionary organizing principles and aid the rational design of both evolutionarily stable and highly evolvable biocircuits. Plasmid copy number and gene circuit design together shape how genetic mutations emerge at the phenotypic level in bacteria. Here the authors characterize how the interplay of gene dosage via plasmid copy number and regulatory architecture affect the phenotypic mutation rate.
|
|
Scooped by
mhryu@live.com
December 31, 2025 9:35 PM
|
Environmental co-contamination presents significant challenges. To tackle these, while microbial consortia offer advantages over single-strain, such as functional redundancy and synergistic degradation, rationally designing effective synthetic microbiomes for complex co-contamination scenarios remains a major challenge. Here, we utilize our advanced genome-scale metabolic modeling (GSMM) tool, SuperCC, to simulate the metabolic behavior of communities consisting of six isolated key strains under single- and multi-carbon source conditions, mimicking single-pollutant or co-contamination scenarios respectively. By integrating multi-omics data with metabolic modeling of cultures, we systematically elucidate key strain interaction networks and adaptive strategies under co-contamination. This reveal that the specific secretory products of broad-spectrum resource-utilizing bacteria serve as key metabolites driving cooperation and highlight the pivotal role of indigenous keystone strains in stabilizing and enhancing community function. Consequently, we propose an innovative and rational paradigm for consortium design: DHP-Com (Degrader-Helper-Potentiator Consortium). Potentiators are top species with stable habitat abundance. Synthetic microbiomes built on this framework exhibit enhanced ecological fitness and substantially improve remediation performance across diverse co-contamination scenarios. Our findings advanced the practical application of GSMM predictions to decipher intricate multi-pollutant/multi-strain interaction networks, offering a powerful rational framework and robust methodological tools for engineering multi-functional and effective synthetic microbiomes for complex environmental remediation. Microbial consortia offer advantages over single strains for the remediation of environmental co-contamination. Here, the authors introduce an Degrader-Helper-Potentiator Consortium framework to design synthetic microbiomes that enhance the remediation of environmental co-contamination.
|
|
Scooped by
mhryu@live.com
December 31, 2025 2:27 PM
|
Vibrio cholerae, the etiological agent of cholera, is ubiquitous in environmental brackish waters. Exposure to low water temperatures induces the bacterium to enter a viable but non-culturable (VBNC) state. In this study, a stepwise decrease in water temperature to 4°C was found to delay the transition to the non-culturable state compared to an abrupt temperature drop, suggesting that V. cholerae cells partially adapt to low temperatures. V. cholerae VBNC cells maintained at 4°C gradually lost their ability to revert to a culturable state. However, VBNC cells in the early stage of dormancy were efficiently resuscitated following treatment with proteolytic enzymes, including proteinase K. The abundance of culturable V. cholerae cells in brackish estuarine waters was quantified using the most probable number (MPN)–quantitative polymerase chain reaction (qPCR) method. Although culturable cells were undetectable in samples treated with bovine serum albumin, they were estimated at 93 and 1,500 MPN/mL in two water samples collected on different days and pre-incubated with proteinase K. Similarly, the abundance of Vibrio species increased markedly following treatment with this enzyme. Additionally, cells of Vibrio species were enumerated by the plating method using CHROMagar Vibrio plates. Consistent with the results of the MPN–qPCR method, treatment with proteinase K resulted in over a 100-fold increase in colony formation. Collectively, these findings suggest that treatment with proteinase K is effective for resuscitating and quantifying V. cholerae VBNC cells in environmental water samples.
|
|
Scooped by
mhryu@live.com
December 30, 2025 3:09 PM
|
RNA-based anti-CRISPRs (Racrs) interfere with the type I-F CRISPR-Cas system by mimicking the repeats found in CRISPR arrays. Here, we determined the cryo-electron microscopy (cryo-EM) structures of the type I-F crRNA-guided surveillance complex (Csy complex) from Pectobacterium atrosepticum and three RacrIF1-induced aberrant subcomplexes. Additionally, we observed that Cas7f proteins could bind to non-specific nucleic acids, forming right-handed superhelical filaments composed of different Cas7 copies. Mechanistically, RacrIF1 lacks the specific S-conformation observed in the corresponding position of the 5′ handle in canonical CRISPR complexes, and it instead adopts a periodic “5 + 1” pattern. This conformation creates severe steric hindrance for Cas5f–Cas8f heterodimer and undermines their binding. Furthermore, Cas7f nonspecifically binds nucleic acids and can form infinite superhelical filaments along Racrs molecules. This oligomerization sequesters Cas6f and Cas7f from binding, therefore blocking the formation of functional CRISPR-Cas effector complexes and ultimately blocking antiviral immunity. Our study provides a structural basis underlying Racrs-mediated CRISPRs inhibition.
|
|
Scooped by
mhryu@live.com
December 30, 2025 2:41 PM
|
Nitrogen availability is a key determinant of plant growth and development. Here, we investigate how different N sources shape Arabidopsis thaliana root system architecture, metabolism and hormone dynamics. L-glutamine (L-GLN) significantly enhances root biomass compared to nitrate (KNO3) without compromising shoot growth. This effect emerges after 2 weeks and is independent of L-GLN's role as a carbon or ammonium source or of potential L-GLN-induced pH changes due to ammonium release, indicating a specific function of L-GLN as a N source and signaling molecule. A reverse genetic screen identified AMINO ACID PERMEASE 1 (AAP1)-mediated uptake and GLUTAMINE SYNTHETASE (GS)-dependent assimilation as essential for L-GLN-induced root biomass. In contrast, the N-sensing regulators NITRATE TRANSPORTER 1.1 (NRT1.1) and AMMONIUM TRANSPORTER (AMT) family members contribute to the differential root responses between KNO3 and L-GLN. Metabolic profiling revealed distinct amino acid signatures under these N sources, irrespective of genotype. Hormonal analyses showed that L-GLN modulates auxin homeostasis, with auxin supplementation restoring primary root growth and lateral root symmetry under L-GLN conditions. L-GLN also reconfigures cytokinin balance by elevating cZ while reducing tZ, collectively shaping root system architecture through hormone-dependent regulation. Together, these findings establish L-GLN as an integrator of N metabolism and hormone signaling in root development, highlighting its signaling capacity beyond nutrient supply and offering new perspectives for improving N use efficiency.
|
|
Scooped by
mhryu@live.com
December 30, 2025 2:26 PM
|
3'-5' translation (termed "backward translation") mediated by eukaryotic circular RNAs has been reported recently. Whether linear mRNA can produce proteins through backward translation remained elusive. Here, we demonstrate that linear mRNAs can mediate backward translation in vitro. Backward translation was detected in human cells transfected with expression vectors used to produce linear mRNAs. Importantly, synthetic linear mRNAs could produce backward translation signal in multiple in vitro translation systems, with a level comparable to that of conventional 5' to 3' forward translation. Furthermore, we demonstrated that a single translation initiating sequence (TIS) was able to drive backward and forward translation in an in vitro experimental setup, with the efficiency of backward translation found to be significant lower than that of forward translation. In summary, this study further reinforces the notion of translation flexibility, demonstrates a broader applicability of backward translation and heralds a new strategy in synthetic biology.
|
|
Scooped by
mhryu@live.com
December 30, 2025 2:10 PM
|
MicroRNAs (miRNAs) are key posttranscriptional regulators of gene expression that influence cancer initiation, progression, and therapeutic response. While most studies have focused on endogenous miRNAs, emerging evidence has highlighted the role of plant-derived miRNAs as exogenous dietary regulators capable of cross-kingdom gene modulation. This review summarises current knowledge regarding plant-derived miRNAs and their ability to regulate human cancer-related genes. Experimental findings indicate that plant miRNAs can withstand gastrointestinal digestion, enter the circulation, and regulate the expression of oncogenes, tumor suppressors, long noncoding RNAs, and immune checkpoint molecules via canonical RNA-induced silencing mechanisms. Specific examples include miR-156a, miR-159a-3p, miR-166a, miR-167e-5p, miR-171, miR-395e, miR-2911, miR-4995 and miR-5754, which exhibit anticancer activities across various cancer types and modulate key signalling pathways in mammalian cells, highlighting their potential as cross-kingdom regulators with therapeutic relevance. In addition to these characterised miRNAs, certain plant groups, which are rich in bioactive compounds, remain unexplored as sources of functional miRNAs, representing a promising avenue for future research. Collectively, these studies underscore the ability of plant-derived miRNAs to modulate mammalian gene expression and suggest their potential as diet-based or synthetic therapeutic agents. Further investigations into their bioavailability, target specificity, and functional relevance could inform innovative strategies for cancer prevention, integrating nutritional, molecular biological, and therapeutic approaches.
|
|
Scooped by
mhryu@live.com
December 30, 2025 11:57 AM
|
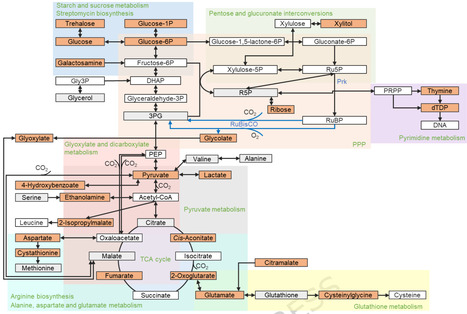
Rising atmospheric CO₂ levels and their impact on climate change have intensified the need for innovative carbon capture and fixation strategies. The Calvin-Benson-Bassham (CBB) cycle, a central metabolic pathway in all photoautotrophic organisms and many autotrophic bacteria, plays a pivotal role in global carbon assimilation but is limited by the low catalytic efficiency of Rubisco. Here, we engineered a complete, functional CBB cycle in Escherichia coli, by heterologously expressing up to 13 genes encoding phosphoribulokinase, α-carboxysomes, and inorganic carbon pumps. This bioengineering approach allowed E. coli to utilize atmospheric CO2 and led to increased levels of sugars such as ribose (4.94-fold) and xylitol (8.94-fold). Detailed metabolomic profiling of central carbon metabolism using gas chromatography-mass spectrometry (GC-MS) demonstrated that installation of the CBB cycle has a notable impact on the metabolic landscape of E. coli, resulting in substantial alterations in central carbon and amino acid metabolism. These findings deepen our understanding of the natural biological carbon-fixation pathway and its engineering in heterotrophic hosts. Furthermore, this work provides a versatile platform for evaluating and selecting efficient carbon-fixation modules, as well as assessing metabolic bottlenecks in engineered systems. These advances offer practical guidance for rational metabolic engineering in diverse organisms for biotechnological applications, including carbon sequestration, sustainable bioproduction, and crop improvement.
|
|
|
Scooped by
mhryu@live.com
Today, 12:59 PM
|
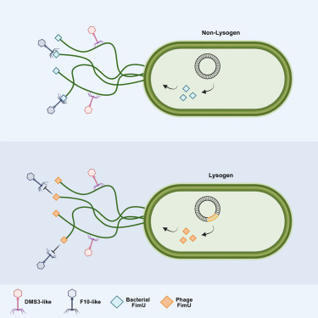
Phage genomes integrated within bacterial genomes, known as prophages, frequently encode proteins that provide defense against further phage infection. These proteins often function by altering the cell surface and preventing phages from attaching to their host receptor. Here, we describe prophage-encoded proteins that resemble FimU, a component of the Pseudomonas aeruginosa type IV pilus. These phage FimU proteins are incorporated into the pilus without altering its function, yet they mediate robust protection against infection by phages that bind to the tip of the pilus, where FimU is located. The phage FimU proteins and the phage tail proteins that likely interact with FimU are highly diverse, suggesting that evolution in this system is driven by phage versus phage competition. These phage FimU proteins represent an example of anti-phage defense mediated by the replacement of a bacterial cell surface component with a phage-encoded protein.
|
|
Scooped by
mhryu@live.com
Today, 11:58 AM
|
Cyanobacteria, as phototrophic organisms with low nutritional requirements and great metabolic versatility, are attractive for the sustainable production of value-added chemicals from CO2 and sunlight. One limitation of these strategies is that carbon is partitioned towards biomass synthesis rather than product synthesis. An alternative to conventional metabolic engineering approaches involves controlling regulatory circuits to enhance the flow of carbon towards the synthesis of desired compounds. The carbon-flow-regulator A (CfrA) is pivotal in redirecting carbon flux during nitrogen deficiency in cyanobacteria, promoting glycogen accumulation by inhibiting 2,3-phosphoglycerate mutase enzyme. The moderately halotolerant cyanobacterium Synechocystis sp. PCC 6803 accumulates sucrose and glucosylglycerol (GG) as compatible solutes under salt stress. Sucrose is a valuable carbon source for heterotrophic organisms, whether they are cultivated independently or in co-cultures. In this context, we explored the potential biotechnological relevance of CfrA in redirecting carbon flow towards sucrose production. A strain that overexpresses cfrA, independently of nitrogen growth conditions, and carries a plasmid that expresses sucrose-phosphate synthase (SPS) from Synechocystis sp. PCC 6803 and the heterologous sucrose permease CscB inducibly (Pars-cfrA/suc strain) was constructed and analysed. In this strain, cfrA expression increased sucrose production by 40% compared to non-induced levels. The fixed carbon was partially redirected towards sucrose production at the expense of glycogen accumulation and biomass generation. Furthermore, an improvement in the photosynthetic activity of this strain was observed due to the presence of this carbon sink. The effect of eliminating GG synthesis (ΔggpS/Pars-cfrA/suc strain) on sucrose production was also analyzed. Under high salinity conditions (400 mM NaCl), this strain exhibited a maximum sucrose accumulation of 2.72 g/L. Encapsulation of the Pars-cfrA/suc strain has also been studied. Our results indicate that modulating carbon flow through CfrA overexpression can substantially boost sucrose production. Glycogen accumulation, mediated by CfrA, enhances sucrose production, which is partly derived from the use of stored glycogen. Furthermore, immobilising Synechocystis cells in alginate improves sucrose production and facilitates its utilisation. Given the widespread occurrence of the cfrA gene in cyanobacteria, its potential as a target in various biotechnological strategies that require the redirection of carbon flow should be considered.
|
|
Scooped by
mhryu@live.com
December 31, 2025 10:24 PM
|
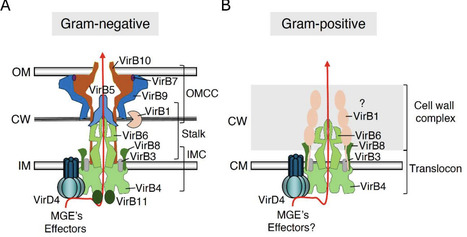
Type IV secretion systems (T4SS) are versatile nanomachines responsible for the transfer of DNA and proteins across cell envelopes. From their ancestral role in conjugation, these systems have diversified into a superfamily with functions ranging from horizontal gene transfer to the delivery of toxins to eukaryotic and prokaryotic hosts. Recent structural and functional studies have uncovered unexpected architectural variations not only among Gram-negative systems but also between Gram-negative and Gram-positive systems. Despite this diversity, a conserved set of core proteins is maintained across the superfamily. To facilitate cross-system comparisons, we propose in this review a unified nomenclature for conserved T4SS subunits found in both Gram-negative and Gram-positive systems. We further highlight conserved and divergent mechanistic and architectural principles across bacterial lineages, and we discuss the diversity of emerging T4SSs whose unique structures and functions expand our understanding of this highly adaptable secretion superfamily..
|
|
Scooped by
mhryu@live.com
December 31, 2025 10:16 PM
|
Phages are viruses that infect bacteria and play essential roles in shaping microbial communities. Identifying phage-host interactions (PHIs) is crucial for understanding infection dynamics and developing phage-based therapeutic strategies. Recent deep learning approaches have shown great promise for PHI prediction; however, their performance remains constrained by the limited number of experimentally validated positive pairs and the overwhelming abundance of unlabeled or non-validated samples. Moreover, most existing models overlook higher-level phylogenetic relationships among hosts, which could provide valuable structural priors for guiding representation learning. To address these challenges, we propose a phylogenetic tree-aware positive-unlabeled deep metric learning framework for phage-host interaction (PHI) identification. Unlike traditional approaches that train classification models to strictly separate positive and negative phage-host pairs, the proposed method learns representations under supervision from both confirmed positive PHIs and host phylogenetic tree constraints on non-positive samples. The proposed method can seamlessly formalize contrastive learning and deep metric learning within the same framework that explicitly optimizes PHI encoders with biological constraints in the learning functions. We show that this metric learning formulation outperforms conventional contrastive learning approaches that enforce separation between positive and negative samples without consistently aligning the learned representations with evolutionary distances. Experiments on the Cherry benchmark dataset and metagenome Hi-C multi-host dataset demonstrate that our approach enhances species-level prediction accuracy, improves cross-host generalization, and yields more interpretable representations of phage-host relationships.
|
|
Scooped by
mhryu@live.com
December 31, 2025 10:02 PM
|
Enzymes do not operate as static structures, but continuously fluctuate between different conformations. Enzymes therefore dynamically sample conformations with varying catalytic activity. However, it remains largely unexplored whether evolution can exploit the conformational dynamics between sub-states to improve activity. Here, we dissect the evolutionary trajectory of the β-lactamase OXA-48 toward improved ceftazidime hydrolysis. Evolution relieved conformational bottlenecks by promoting alternate functional sub-states, gradually shifting the rate-limiting step from substrate binding to sub-state interconversion, and finally to the chemical step. Reorganization of the conformational landscape enhanced OXA-48's ability to hydrolyze ceftazidime and introduced a trade-off in its native activity against meropenem. This trade-off stemmed from catalytic incompatibility between the native and the evolved sub-state populations. Our findings highlight the transitions between functional sub-states as a mechanism of natural selection, shaping functional divergence and offering new strategies for enzyme and antibiotic engineering.
|
|
Scooped by
mhryu@live.com
December 31, 2025 9:42 PM
|
Phage engineering holds significant potential for overcoming the challenges that limit phage therapy. A promising yet underutilized approach is engineering phage host range to target specific receptors and guiding bacterial evolution into a desirable trajectory, referred to as evolutionary steering. This requires balancing binding interactions between native and desired receptors, though it is unclear how balancing binding interactions affects host range and resulting evolutionary trajectories. To provide insights, we surveyed a suite of phage engineering methods to program protein-protein interactions between phage and bacteria and then measured host range expansion and evolutionary trajectories. We engineered T3 and T7, both LPS-targeting phages, to target a proteinaceous nanobody receptor. In addition, we discovered that the capsid plays a role in phage host range and can be a potential target for host range expansion. Together, these studies increase the therapeutic potential for T3 and T7, and more broadly for LPS-targeting phages.
|
|
Scooped by
mhryu@live.com
December 31, 2025 2:37 PM
|
Cells combat peroxide stress using peroxiredoxins and catalases. The paradigmatic H2O2-sensing OxyR or PerR transcription regulators typically control their expression in bacteria. Here, we report our discovery of a noncanonical mechanism for H2O2-signaled regulation of peroxiredoxin ahpC and catalase katB genes in Myxococcus xanthus by PexR, an ATP-binding bacterial enhancer binding protein that acts as a dual-function repressor-activator. PexR, a dimer capable of higher-order oligomerization, binds to dyad-symmetry repeats upstream of ahpC and katB, activating their H2O2-induced σ54-dependent expression. Under peroxide stress-free conditions, PexR downregulates housekeeping σA-dependent ahpC expression. Deleting ahpC causes pleiotropy, and synthetic lethality when eliminated with katB or pexR, indicating PexR-mediated co-regulation of AhpC and KatB as critical for normal growth. We show that resting state PexR is autoinhibited by its N-terminal Zn2+/Fe2+-binding GAF (cGMP-specific phosphodiesterases, adenylyl cyclases, and FhlA) domain, which senses H2O2 and releases its bound metal to trigger PexR-activated σ54-dependent expression. Our genomic analyses reveal conservation of PexR and its regulatory elements and, likely, mechanism across Myxococcota, frequently co-occurring with OxyR or PerR, showcasing this phylum’s remarkable diversity of potential peroxide stress response regulatory mechanisms. Moreover, PexR likely operates in phyla beyond Myxococcota, and its discovery expands the toolkit for genetically encoded H2O2 sensors.
|
|
Scooped by
mhryu@live.com
December 30, 2025 3:28 PM
|
Argonaute proteins provide innate immunity in all domains of life through guide-dependent recognition and cleavage of invader nucleic acids. Many short prokaryotic Argonautes (pAgos) lack nuclease activity and are instead co-encoded with tentative nuclease effectors, but their activation mechanisms remain unknown. Here, we characterize SPARHA systems (short prokaryotic argonautes, HNH-associated), containing HNH nuclease effectors. RNA-guided target DNA recognition by pAgo induces formation of SPARHA filaments with a chain of double nuclease sites formed by HNH tetramers, leading to indiscriminate collateral degradation of DNA and protecting bacterial population from invaders. We show that the assembly of filaments proceeds via a universal activation pathway involving a cascade of target-induced conformational changes in SPARHA, conserved in other short pAgo systems containing various types of effectors. pAgos and associated effectors act as modular defense systems that translate recognition of specific DNA into immune response through assembly of supramolecular complexes, deleterious for invaders and potentially useful for biotechnology. Argonaute proteins degrade specific invader nucleic acids in eukaryotic and prokaryotic innate immunity. Here, Kanevskaya et al. describe a bacterial immune system in which RNA-guided recognition of invader DNA by Argonaute triggers formation of HNH nuclease filaments with collateral activity, protecting the bacterial population from invaders.
|
|
Scooped by
mhryu@live.com
December 30, 2025 2:56 PM
|
Natural CRISPR-Cas9 systems provides diverse properties for genome editing, yet finding compact variants remains a priority. In this study, we screened a panel of 11 CjCas9 orthologous using a GFP activation assay and identified seven active nucleases. Among these, Cj4Cas9 stood out as particularly noteworthy due to its compact genome size (985 amino acids) and unique PAM preference (5’-NNNGRY-3’). Cj4Cas9 demonstrates efficient disruption of the Tyr gene in mouse zygotes, resulting in an albino phenotype. Furthermore, when delivered via AAV8, Cj4Cas9 achieves efficient genome editing of the Pcsk9 gene in mouse liver, leading to reduced serum cholesterol and LDL-C levels. Seeking to further expand its utility, we engineered Cj4Cas9 for higher activity by introducing L58Y/D900K mutations, resulting in a variant termed enCj4Cas9. This variant exhibits a two-fold increase in nuclease activity compared to the wild-type Cj4Cas9 and recognizes a simplified N3GG PAM, considerably expanding its targeting scope. These findings establish Cj4Cas9 and its engineered variants for fundamental research and therapeutic applications. A compact Cas9 ortholog, Cj4Cas9, enables efficient genome editing in vivo compatibility.
|
|
Scooped by
mhryu@live.com
December 30, 2025 2:35 PM
|
With the rise in antimicrobial resistance, understanding the virulence factors utilized by pathogenic E. coli is essential for the development of alternative therapeutics. While previous work has shown that disruption of the E. coli rhomboid protease gene glpG leads to defects in bacterial colonization, here we provide mechanistic insight into the loss of fitness. We show GlpG is essential for the assembly of type 1 pili, a virulence factor required for the colonization of eukaryotic cells. Since pili are critical for biofilm formation and bacterial persistence, the absence of GlpG proteolytic activity reduces the production of biofilm. Working towards new potential antimicrobial targets for treating infections, we show that biofilm formation is hampered by GlpG inhibition. Our data demonstrates that GlpG plays a key role in protein quality control of type 1 pili and alters the paradigm for GlpG proteolysis, previously implicated in the cleavage of only membrane embedded substrates. The disruption of the rhomboid protease gene glpG in E. coli leads to defects in bacterial colonization. Here, the authors show that GlpG is essential for the assembly of type 1 pili, a virulence factor required for colonization of bacteria in eukaryotic cells.
|
|
Scooped by
mhryu@live.com
December 30, 2025 2:22 PM
|
Droplet microfluidics is a core technology that powers high-throughput single cell sequencing. However, the current generation of single-cell microfluidics faces notable limitations, including cell aggregation, suboptimal on-chip reactions that compromise experimental outcomes and elevate background noise, as well as a dependence on costly commercial barcode beads. To address these challenges, we present MusTer, an integrated next-generation platform with multi-parametric singlet droplet sorting and triple-droplet merging capability. MusTer's multi-parametric singlet sorting module enables in-line droplet analysis of intrinsic fluorescence peak amplitude, width and interval from single-nucleus- (singlet) or multiple-nuclei (multiplet)-encapsulating droplets, subsequently allowing an effective separation of the singlet droplets from multiplet droplets and empty droplets. MusTer's triple-droplet merging module enables precise multi-step reactions, with each step performed under its own optimal conditions, thereby significantly enhancing experimental flexibility and efficiency. We validated MusTer's performance by performing single-cell ATAC-seq on maize leaves. The results demonstrate that MusTer significantly reduces the doublet rate, enhances the signal-to-noise ratio, and yields improved cell clustering compared with traditional methods. These results validate MusTer's capability to overcome key limitations in droplet-based single-cell analysis, effectively enhancing data quality and reliability, and also paves the way for its use in other challenging sample types and multi-step single-cell assays.
|
|
Scooped by
mhryu@live.com
December 30, 2025 1:16 PM
|
Temperature offers a simple yet powerful signal to program cellular behavior. Here, we engineered and characterized a set of temperature-dependent genetic circuits that integrate RNA thermometers with site-specific DNA recombinases to achieve precise, irreversible control of gene expression. Using the serine recombinase Bxb1 placed under the control of the Salmonella FourU RNA thermometer, we demonstrate how promoter strength critically defines thermal sensitivity: weak promoters’ activity clears ON/OFF transitions, while strong promoters lead to continuous, quasi-temperature-independent recombination. Furthermore, temperature pulse duration and growth phase of cell culture were found to modulate recombination efficiency, providing additional layers of control. We illustrate the potential of this framework through proof-of-concept applications, including (i) the generation of spatial expression patterns on 2D surfaces via localized heating, (ii) a paper-based device capable of recording temperature gradients as stable genetic outputs, and (iii) a temperature-triggered lysis system for controlled cellular release. Together, these results establish temperature-regulated recombinase circuits as versatile and robust tools for programmable, spatially resolved, and irreversible control of gene expression, paving the way for new applications in synthetic biology, biosensing, and bioproduction.
|


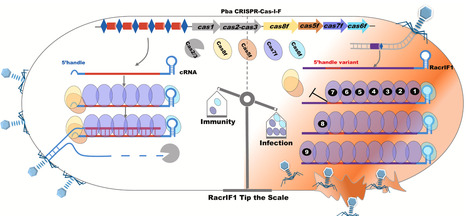


























Racrs bind Cas proteins to form aberrant CRISPR complexes, thus interfere with the type I-F CRISPR-Cas system, thereby preventing their participation in anti-phage defense.