 Your new post is loading...

|
Scooped by
mhryu@live.com
December 29, 11:34 PM
|
Biomaterials are gaining attention as sustainable alternatives in the fashion industry. In a recent Trends in Biotechnology article, Zhou et al. optimized a one-pot strategy for the growth of colored bacterial cellulose, enabling more sustainable production of textile materials. Here, we contextualize this development alongside other innovations and roadblocks in sustainable fashion.
|
|
Scooped by
mhryu@live.com
December 29, 10:46 PM
|
Design of experiments (DOE) principles are increasingly applied to biological assays, yet it remains unclear whether their foundational assumption, orthogonal decomposition, holds in nonlinear biological systems. We addressed this question using Perturb-seq as a case study. By benchmarking a design commonly used in Perturb-seq and related experiments against the orthogonal Plackett-Burman (PB) design via simulations, we uncovered a counter-intuitive phenomenon: while orthogonal designs generally excel, the multicollinearity inherent to the common design is functionally advantageous in systems with significant signal amplification. This challenges the blind application of DOE to biology. Based on these findings, we developed the PB suitability index (PBSI), a simple, parameter-free metric that predicts the optimal design solely from network structure. Our work not only provides practical guidelines for Perturb-seq but also establishes a "biology-oriented DOE" framework, bridging the gap between statistical rigor and biological complexity.
|
|
Scooped by
mhryu@live.com
December 29, 10:39 PM
|
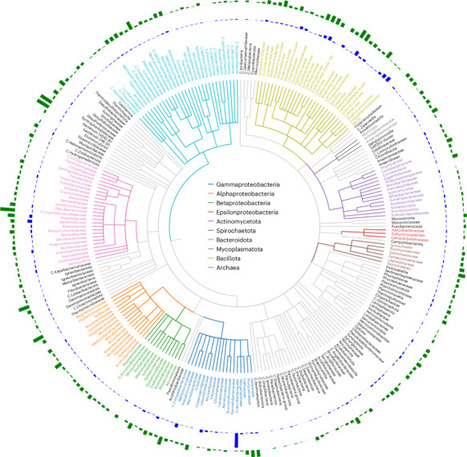
Microorganisms underpin global biogeochemical cycles and represent a vast, underexplored reservoir for sustainable biotechnology and human health. Their diversity, arising from the interplay of numerous genes within holistic genomic contexts, dictates complex ecophysiological traits across varied environments. To bridge the gap between complex genomic sequences and associated biological functions and phenotypic outcomes, we present MicroGenomer, a foundation model for transferable microbial genome representations enabling multi-scale genomic understanding and ecophysiological trait prediction. MicroGenomer leverages a hierarchical training strategy comprising pre-training on large-scale genomic sequences (234.5 billion base pairs), domain-specific mid-training using the GTDB-curated marker gene set, and task-specific post-training. Despite a streamlined architecture of 470 million parameters, MicroGenomer achieves performance on key tasks competitive with models nearly 85 times its size. Advancing beyond established gene-scale encoders, MicroGenomer generates robust embeddings at the genome scale for downstream modeling. Extensive evaluations demonstrate that MicroGenomer effectively captures phylogenetic structures in species space, excelling in gene- and genome-scale understanding as well as ecophysiological trait prediction. The practical utility of these capabilities is further demonstrated by targeted wet-lab validation on newly isolated strains, indicating that MicroGenomer's predictions provide reliable guidance for biological experiments. Collectively, MicroGenomer offers a high-performance, resource-efficient framework that transforms raw sequence data into actionable biological insights, providing a powerful foundation for microbiome research and biotechnology.
|
|
Scooped by
mhryu@live.com
December 29, 3:03 PM
|
Antimicrobial peptides (AMP) are crucial in addressing the global crisis of bacterial resistance. However, there are still significant limitations in existing methods on de novo AMPs design, especially in designing AMPs with desirable physicochemical properties for specific bacterial pathogens. In this study, we propose a novel generative framework for designing pathogen-targeted antimicrobial peptides with programmable physicochemical properties. More specifically, a conditional Variational Autoencoder is first pretrained for generating AMPs with editable physicochemical properties. We then develop a conditional diffusion model to learn hidden representations of AMPs for targeting pathogens of interest, and construct corresponding MIC predictors for specific bacterial strains. Through comprehensive simulation experiments, we demonstrate that the proposed framework outperforms most existing models in terms of antimicrobial efficacy against specific bacterial targets. Moreover, through systematic screening and analysis, we have identified two star AMPs for each of the two target bacterial species (i.e., E. coli or S. aureus), both of which exhibit excellent performance in antibacterial activity, hemolytic properties, toxicity profiles, etc. Overall, this study provides the key technological support for developing next-generation intelligent platforms for antimicrobial agents design.
|
|
Scooped by
mhryu@live.com
December 29, 2:55 PM
|
Phages are typically classified as temperate, integrating into host genomes, or lytic, replicating and killing bacteria; for this reason, lytic phages are not expected in bacterial genome sequences. Here we analyse 3.6 million bacterial genome assemblies from 1,226 species and find 119,510 lytic phage genomes, which we term bacterial assembly-associated phage sequences. This represents a ~5-fold increase in the number of phages with associated hosts and raises questions about fundamental aspects of phage biology. Our analyses of bacterial assembly-associated phage sequences revealed previously undescribed phage clusters, including clusters distantly related to Salmonella Goslarviruses in Escherichia coli and Shigella, while also substantially expanding known genera such as Seoulvirus (from 16 to >300 members). Close relatives of lytic phages used therapeutically were also detected, suggesting clinical isolate sequencing unknowingly archives potential phage candidates. The discovery of complete, lytic phage genomes within bacterial assemblies challenges assumptions about the nature of the lytic lifestyle and reveals an untapped reservoir of phages. Diverse genomes of lytic phages are found in bacterial assemblies, challenging assumptions about the nature of the lytic lifestyle.
|
|
Scooped by
mhryu@live.com
December 29, 2:48 PM
|
Untargeted, also known as metagenomic, nanopore sequencing is a powerful tool for virus genomic surveillance, particularly in resource-limited settings and when paired with the portability of the MinION device (Oxford Nanopore Technologies, ONT). However, a major bottleneck for global access is the absence of a user-friendly software capable of efficiently analyzing untargeted nanopore sequencing data to generate high-quality consensus genomes. We share ViMOP, a pipeline built on our long-term experience in nanopore field sequencing. The pipeline emphasizes field user-friendliness, flexibility and versatility to analyze reads generated directly from human clinical samples. The software assembles de novo contigs, matches contigs to known viral references and uses them to assemble consensus genomes. Executed with a single Nextflow command or via the EPI2ME Desktop interface (ONT), results are summarized in an HTML report. ViMOP, through its user-centered design, lowers the barrier to high-quality virus genome reconstruction and advances capacity for genomic surveillance.
|
|
Scooped by
mhryu@live.com
December 29, 2:30 PM
|
Sporulation is a strategy employed by many bacteria to survive harsh environmental conditions. The genus Paenibacillus includes spore-forming species notorious for spoiling pasteurized dairy products and for causing American foulbrood in honeybee larvae, leading to colony collapse. Human pathogens within Paenibacillus are also a growing threat, causing fatal opportunistic infections. Here, we present a comprehensive survey of sporulation genes across 1,460 high-quality Paenibacillus genomes. We find that all members of the sporulation-initiating phosphorelay are well conserved, but that the Spo0B phosphotransferase contains a predicted transmembrane domain. We confirm that this domain localizes Spo0B to the cell membrane and therefore refer to this Spo0B variant as Spo0B-TM. Spo0B-TM is present in 92% of surveyed Paenibacillus genomes. Consistent with its high level of conservation, we find that the transmembrane domain is important for detecting its interaction with its phosphorelay partners Spo0A and Spo0F. Moreover, we find that Spo0B exhibits low sequence identity across Bacillota when compared with other members of the phosphorelay. Altogether, this work highlights the potential for diversity even within the highly conserved phosphorelay that initiates sporulation in Bacillota.
|
|
Scooped by
mhryu@live.com
December 29, 2:23 PM
|
AlphaFold (AF) has transformed protein structure prediction and is increasingly enabling the rational redesign of glycosyltransferases (GT) involved in the biosynthesis of human milk oligosaccharides (HMOs). AF-derived structural insights deepen our understanding of GT structure–function relationships, supporting targeted engineering to enhance the catalytic efficiency, broaden substrate scope, and improve regiospecificity─key requirements for scalable microbial production of complex long-chain HMOs. Using lacto-N-fucopentaose I (LNFP I) synthesis as a case study, we illustrate how AF-guided modeling facilitates GT redesign for efficient HMO biosynthesis, while also discussing current limitations and challenges toward precision GT engineering.
|
|
Scooped by
mhryu@live.com
December 29, 2:09 PM
|
Antimicrobial resistance continues to rise globally, with biofilm-associated infections intensifying the clinical burden through persistent tolerance to antibiotics and evasion of immune responses. Biofilms, structured microbial communities embedded in a protective extracellular matrix, underlie many chronic and recurrent infections, including endocarditis, urinary tract infections, cystic fibrosis lung disease, and device-related infections. Conventional antibiotics often fail in these contexts, and the discovery pipeline for novel agents remains limited. Nano-technology has therefore emerged as a promising alternative, offering unique physicochemical features that enable enhanced penetration into biofilm matrices, improved drug stability, and targeted delivery of therapeutic agents. Diverse nanosystems, including metallic, polymeric, lipid-based, and ligand-functionalized platforms, have shown encouraging results in vitro and in vivo, demonstrating superior biofilm disruption and bacterial eradication compared with conventional therapies. Nevertheless, translating these advances into clinical practice remains challenging. Key barriers include complex and costly synthesis, scalability under good manufacturing practices, limited drug loading efficiencies, variability of preclinical biofilm models, regulatory uncertainties, and the risks of nanoparticle (NP)-induced toxicity, unpredictable biodistribution, and potential resistance development. Moreover, the dynamic interactions between NPs, host fluids, and biofilm extracellular matrices complicate pharmacokinetic and pharmacodynamic predictability. Addressing these obstacles requires coordinated efforts to refine manufacturing processes, standardize biofilm models, and implement nanospecific regulatory frameworks. With careful optimization, nanomedicine holds the potential to redefine the therapeutic landscape for biofilm-related infections.
|
|
Scooped by
mhryu@live.com
December 29, 1:22 PM
|
Understanding microbial phenotypes from genomic data is crucial for studying co-evolution, ecology, and pathology. This study presents a scalable approach that integrates literature-extracted information with genomic data, combining natural language processing and functional genome analysis. We applied this method to publicly available data, providing novel insights into predicting microbial phenotypes. We fine-tuned transformer-based language models to analyze 3.83 million open-access scientific articles, extracting a phenotypic network of bacterial strains. This network maps relationships between strains and traits such as pathogenicity, metabolism, and biome preference. By annotating their reference genomes, we predicted key genes influencing these traits. Our findings align with known phenotypes, reveal novel correlations, and uncover genes involved in disease and host associations. The network’s interconnectivity provides deeper understanding of microbial communities and allowed identification of hub species through inferred trophic connections that are difficult to infer experimentally. This work demonstrates the potential of machine learning for uncovering cross-species gene–phenotype patterns. As microbial genomic data and literature expand, such methods will be essential for extracting meaningful insights and advancing microbiology research. In summary, this integrative approach can accelerate discovery and understanding in microbial genomics. Ultimately, such techniques will facilitate the study of microbial ecology, co-evolutionary processes, and disease pathogenesis to an unprecedented depth.
|
|
Scooped by
mhryu@live.com
December 29, 1:11 PM
|
Acid resistance is crucial for enterobacteria to withstand host acidic environments during infection, including the gastrointestinal tract and macrophage phagosomes. A key acid resistance mechanism of the facultative intracellular pathogen Salmonella is the expression of the arginine decarboxylase AdiA. While AdiA confers acid resistance via an H+-consuming reaction, we discover that the 3′-untranslated region (UTR) of adiA mRNA is processed by RNase E into a regulatory small RNA, AdiZ. Through RNA–RNA interactome profiling and transcriptomic analysis, followed by in vitro structural probing and in vivo validations, we demonstrate that AdiZ directly base-pairs with and negatively regulates ptsG, pykF, and dmsA mRNAs involved in glucose uptake, glycolysis, and anaerobic respiration, respectively. Intriguingly, AdiZ is induced and facilitates Salmonella survival within macrophages, where acidic and hypoxic stresses prevail. Thus, simultaneous expression of AdiA and AdiZ from a single mRNA ties arginine-dependent acid resistance to metabolic reprogramming of Salmonella in the host intracellular niches.
|
|
Scooped by
mhryu@live.com
December 29, 11:59 AM
|
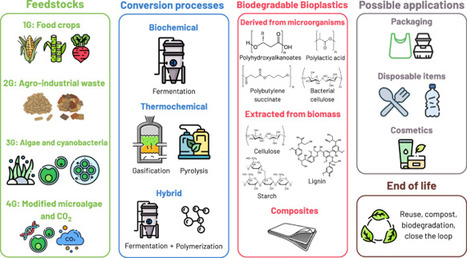
Biodegradable bioplastics have emerged as promising alternatives to conventional plastics in the current scenario of growing demand for sustainable materials. However, the high costs associated with their production still interfere with the proper dissemination of these materials. The present review will deal with the different aspects of the production of biodegradable bioplastics in biorefineries as an approach for cost reduction and low waste generation, aligning with circular bioeconomy principles. By employing different types of biomass and conversion processes, bioplastics and their composites can be considered a valuable product in biorefineries, demonstrated by actual case studies and functional industries.
|
|
Scooped by
mhryu@live.com
December 28, 12:42 PM
|
To address the growing emergence of multi-resistant phytopathogenic bacteria, innovative solutions are being explored in the field of plant health. Among them, bacteriocins, antimicrobial peptides or proteins secreted by bacteria, characterized by a highly specific spectrum of activity and involved in intra-specific competition, are gaining increasing interest. Bacteriocins can confer a positive selective advantage in both natural and agricultural environments, thereby contributing to microbiome modulation. Bacteriocin-producing rhizobacteria and lactic acid bacteria are already used as biocontrol agents against phytopathogenic bacteria, as well as plant growth stimulators. Bacteriocins can be produced in situ by using avirulent strains, or ex situ through industrial synthesis and applied as biopesticides. Nowadays, genetic engineering enables production of chimeric bacteriocins and their direct production in transgenic plants, avoiding the need for repeated treatments and limiting emergence of resistances. The selection of promising bacteriocins can be guided by omics-based approaches, notably metagenomics, which involve the direct extraction and sequencing of DNA from environmental samples and provides access to the genetic diversity in complex soil or plant-associated microbiomes. Combined with open-access databases and recently developed integrated tools, this approach not only facilitates the identification of known structures of bacteriocins, but also enables the prediction of potentially active peptides even those never experimentally characterized. Bacteriocin-based strategies, at the crossroads of molecular biology, microbial ecology and agronomy, hold significant potential for promoting sustainable agriculture through highly specific pathogen targeting. However, their large-scale implementation still faces several challenges, including standardization of strain screening protocols, compliance with regulatory frameworks and farmer acceptance.
|
|
|
Scooped by
mhryu@live.com
December 29, 10:55 PM
|
Solanoeclepin A (SolA) is a triterpenoid exuded from the roots of Solanaceae plants, originally identified as hatching stimulant for plant parasitic cyst nematodes. Assuming that evolution would have selected against the production of such a fitness lowering molecule, we postulated that SolA must serve another, beneficial, role for the plant. In this study, we demonstrate that nitrogen (N) deficiency strongly increases the SolA concentration in tomato root exudate. Moreover, SolA is produced only under non-sterile conditions, indicating that soil microbiota are involved in its production. Time-resolved RNAseq analysis revealed several candidate genes for SolA biosynthesis, which were all upregulated under N deficiency. Transient silencing of two SolA biosynthetic genes (e.g., CYP749A19 and CYP749A20) significantly reduced SolA production. Microbiome analysis on the rhizosphere of these plants demonstrated that the recruitment of beneficial Massilia spp. was inhibited in the transiently silenced plants. Isolation of a Massilia strain, identified as Massilia cellulosiltytica, allowed us to show that it has growth-promoting activity under N deficiency, likely via indole-3-acetic acid production and enhanced N acquisition. Root exudate of N-starved tomato displayed strong chemotactic activity towards this strain. Together, these findings demonstrate that SolA production under N deficiency relies on the interaction between tomato and soil microbiota that convert a plant-produced precursor that is a recruitment signal for beneficial, growth-promoting microbes to SolA, which was hijacked by cyst nematodes as a reliable host presence cue. This dual functionality resembles the microbial transformation of primary into secondary bile acids, important signalling molecules, in the gut of animals, and suggests convergent evolution in host microbe co-metabolism. Overall, our study positions SolA as a multifunctional signaling molecule in rhizosphere interactions, with potential application for enhancing crop resilience and sustainable pest management.
|
|
Scooped by
mhryu@live.com
December 29, 10:43 PM
|
The identification of antifungal proteins (AFPs) is crucial for understanding microbial interactions and facilitating the discovery of antifungals on a large scale from genomic and metagenomic data. Most existing computational tools are developed for predicting antifungal peptides rather than proteins. To address this limitation, we developed AntiFP2. A manually curated dataset of experimentally validated AFPs was used to train and develop prediction models using cross-validation techniques. The ensemble strategy, which combined ESM2, BLAST, and MERCI, achieved the best results across independent validation. Beyond prediction, we implemented a complete pipeline for genome- and metagenome-wide AFP screening. AntiFP2 is freely available as a web server, standalone package, and Docker container, enabling scalable and reproducible antifungal protein discovery.
|
|
Scooped by
mhryu@live.com
December 29, 3:07 PM
|
Heterotrophic nanoflagellates are the chief agents of bacterivory in the aquatic microbial loop but remain underrepresented in culture collections and in genomic databases. We isolated and characterized a representative of the previously uncultured freshwater Cryptomonad Group 1 (CRY1a) lineage using a genome-streamlined, ultra-small and abundant microbe Planktophila versatilis as a prey and Catalyzed Reporter Deposition-Fluorescence in situ Hybridization (CARD-FISH) probe–based screening. This isolate, Tyrannomonas regina, is one of the most dominant ubiquitous heterotrophic cryptomonads in freshwaters. It is a small heterotrophic nanoflagellate (ca. 3–5 μm) and has the smallest genome of any cryptomonad sequenced thus far. The compact genome (ca. 69 Mb) revealed no traces of a photosynthetic lifestyle, consistent with its phylogenomic placement as a sister clade to cryptophytes that are characterized by the acquisition of a red-algal symbiont. Moreover, in comparison to its photosynthetic counterparts, its genome presents substantially lower repeat content and endogenous viral elements. Genomes of two giant viruses, Tyrannovirus reginensis GV1 and GV2, were also recovered from the same culture and represent a viral genus that has been described so far solely by metagenome-recovered genomes. Collectively, these findings provide insights into genomic ancestry and evolution, widespread ecological impact, and interactions of an elusive protist lineage and illustrate the advantages of culture-centric approaches towards unfolding complex tapestries of life in the microbial world.
|
|
Scooped by
mhryu@live.com
December 29, 3:01 PM
|
Coordinating multiple liquid handling robots is a complex logistical task when designing biological experiments. Protocol designers must consider the capabilities and constraints of each robot to distribute work optimally across multiple instruments. We developed an optimization framework that finds optimal liquid handling solutions that leverage an arbitrary number of robots. Our algorithm, called Pourfecto, abstracts the capabilities of each robot and their labware, allowing us to plan and schedule a wide range of biological experiments using commercial instruments and custom-built hardware. Pourfecto can optimize multiple objectives (minimum transfers, fewest reagents, fewest labware swaps) and scales to experiments with hundreds of thousands of liquid transfers.
|
|
Scooped by
mhryu@live.com
December 29, 2:51 PM
|
Bacteria have a variety of mechanisms for limiting predation by phages. SpbK is a Toll/interleukin-1 receptor (TIR) domain-containing antiphage defence protein from Bacillus subtilis that provides protection against the temperate phage SPβ via abortive infection. Here we structurally characterise SpbK and its interaction with the SPβ protein YonE. We demonstrate that SpbK is an NADase that produces both ADP-ribose (ADPR) and canonical cyclic ADPR with a N1-glycosidic bond (cADPR, also referred to as N1-cADPR). Combining cryo-EM, in silico predictions, site-directed mutagenesis, and phage infection assays, we show that formation of two-stranded head-to-tail assemblies of SpbK TIR domains is required for both NADase activity and antiphage defence. We also demonstrate that YonE is a dodecameric portal protein that activates the NADase function of SpbK by facilitating TIR domain clustering. Collectively, our results provide insight into how bacterial TIR NADases recognise phage infection. SpbK protects Bacillus subtilis from phage infection by depleting NAD⁺. In this study, the authors uncover the molecular mechanisms underlying SpbK’s self association-dependent NADase activity and its activation by the SPβ phage portal protein YonE.
|
|
Scooped by
mhryu@live.com
December 29, 2:41 PM
|
Microbial consortia show promise for bioremediation of environmental pollution, but performance optimization and risk assessment remain challenging due to unculturable species and limitations of traditional biochemical and sequencing tools. This study demonstrates how a multi-omics approach can provide deeper insight into the performance and risks of using a model aerobic ammonia-oxidizing consortium under conditions representative of wastewater treatment. Long-read DNA sequencing recovered several high-quality genomes, revealing dominance by an unclassified Nitrosospira species with expected ammonia oxidation capabilities. Lower-abundance taxa with nitrogen cycling potential were also detected, though species-level identification was limited by poor taxonomic database representation. Multi-omics and nitrogen analyses showed shifts in community composition and nitrogen cycling activity when the consortium was grown along a redox gradient typical of wastewater. All cultures accumulated ammonia over 4 weeks, with only aerobic cultures reducing ammonia levels thereafter. The dominant Nitrosospira population declined in abundance and activity in aerobic cultures while shifting toward nitrogen reduction under anoxic conditions. This metabolic shift would not have been detected using amplicon sequencing alone. Multi-omics also supported risk assessment through detection of waterborne pathogens from the Legionella genus and other lineages harboring virulence genes resembling those from known pathogens. This study highlights the value of multi-omics for optimizing microbial consortia and assessing biosafety risks but also underscores challenges related to effective data analyses and the feasibility of risk assessment under realistic conditions. Addressing these challenges will be essential to support the broader adoption of multi-omics strategies by stakeholders working with microbial consortia across diverse environmental applications.
|
|
Scooped by
mhryu@live.com
December 29, 2:28 PM
|
Methane emissions from rice paddies represent a critical environmental concern in agriculture. Although genetic strategies for mitigating emissions have gained attention, the specific microbial and molecular mechanisms remain underexplored. Here, we investigated how the gs3 loss-of-function allele in the near-isogenic rice line Milyang360 modulates rhizosphere and endosphere microbial communities under distinct nitrogen regimes. Field experiments revealed that Milyang360 consistently reduced methane emissions compared with its parental line, Saeilmi, particularly under low-nitrogen conditions. Integrated plant transcriptomic and rhizosphere metagenomic analyses, including the reconstruction of Metagenome-Assembled Genomes, demonstrated that the gs3 allele upregulated genes related to root hair elongation or promoting microbial symbiosis. This physiological change limited substrate availability for methanogens and facilitated the colonization by beneficial microorganisms. Consequently, we observed a functional shift in the microbiome, characterized by the enrichment of methanotrophs and nitrogen-fixing bacteria. This microbial restructuring was most prominent under low-nitrogen conditions, indicating a strong genotype by environment interaction. Our findings highlight the gs3 allele’s dual role in reducing methane emissions and improving nitrogen use efficiency by recruiting a beneficial microbiome. This study provides a clear mechanistic link between a plant gene and rhizosphere ecology, offering a promising genetic target for developing sustainable, low emission rice cultivars.
|
|
Scooped by
mhryu@live.com
December 29, 2:13 PM
|
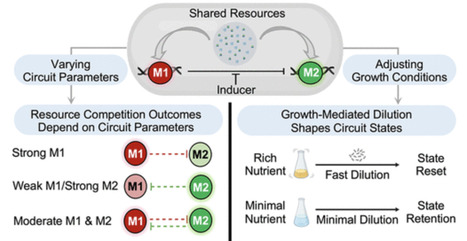
In synthetic biology, a key goal is to design robust and stable genetic circuits with accurate and predictable behavior. Modularity is a central principle in circuit design, enabling the construction of complex systems from smaller, well-characterized parts. However, resource competition presents a major obstacle to modularity by disrupting gene expression dynamics. Here, we constructed and characterized a library of inhibitory genetic cascades with varied promoter strengths, RBS strengths, and plasmid backbones. We found that increasing the expression of the downstream module could unexpectedly lead to a reduction in the expression of the upstream regulatory module by competing for shared cellular resources. These results indicate that resource limitations can transform a unidirectional inhibitory cascade into an unintended feedback loop. In addition, we found that growth-mediated dilution can reshape gene expression patterns, further influencing circuit dynamics. Together, these findings underscore the critical roles of both resource competition and growth dilution in shaping the behavior of synthetic gene circuits.
|
|
Scooped by
mhryu@live.com
December 29, 1:25 PM
|
High expression of heterologous proteins is often achieved by integrating multiple copies of a gene into a host. However, such multicopy systems are prone to genetic instability due to homologous recombination between identical sequences. We present the multisequence ChimeraMap (MScMap), an algorithm for designing multiple synonymous coding sequences that minimizes recombination risk while maintaining high expression. MScMap extends the ChimeraMap framework by selecting diverse nucleotide blocks from a host genome to encode the target protein, balancing host adaptation and sequence dissimilarity. We introduce heuristics for block selection and concatenation to reduce long common substrings, a known driver of recombination. Our method outperforms a multi-objective evolutionary algorithm in both genetic stability and predicted expression across a wide range of human proteins while being significantly faster. We also show that MScMap can also be used to reduce sequence repeats within a single coding sequence. A web tool for single and multicopy coding sequence optimization is available online.
|
|
Scooped by
mhryu@live.com
December 29, 1:17 PM
|
This work aims to improve RNA synthesis and manufacturing, exemplified by T7 RNA polymerase-driven in vitro transcription. We developed a novel, plasmid-compatible co-tethering strategy that functionally couples RNA polymerase to its promoter DNA immobilized on a solid matrix. As demonstrated recently, co-tethering enhances promoter binding, increases RNA yield, and suppresses RNA re-binding, especially under high-salt conditions, thereby reducing double-stranded RNA by-products. The system leverages asymmetric end-labeling of linearized plasmid DNA using a simple “Klenow fill-in” reaction with modified nucleotides, enabling stable attachment of DNA to both RNA polymerase and solid support (magnetic beads). The immobilized co-tethered polymerase–DNA complex supports efficient transcription initiation in high-salt environments (which further reduces RNA re-binding), yielding RNA of high purity. Co-tethered complex remains functionally stable over extended storage and multiple transcription cycles (10-20 rounds), re-using the enzyme–DNA catalyst. Transcripts of lengths (0.8, 5.6, and 8.6 kb) are efficiently produced. Highly sensitive in vitro assays with immune cells confirm low immunogenicity and strong translational output, while in vivo validation using a novel Matrigel-plugged mouse model demonstrates robust expression and safety. With a simple modification to the DNA template, the reusable, co-tethered enzyme–DNA catalytic complex streamlines mRNA manufacturing by producing RNA of higher purity from the outset.
|
|
Scooped by
mhryu@live.com
December 29, 12:56 PM
|
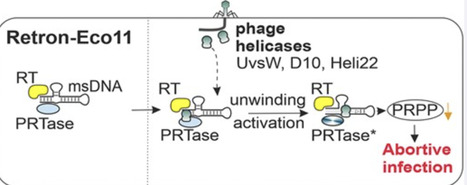
Retrons are tripartite bacterial systems composed of non-coding RNA (ncRNA), reverse transcriptase (RT), and effector proteins with diverse enzymatic domains. Here, we characterized Retron-Eco11, a type III-A3 retron associated with a phosphoribosyltransferase-like effector, and demonstrated that it mediates antiphage defense and can be harnessed for genome editing. All three components—ncRNA, RT, and effector—are essential for defense. Retron-Eco11 protects Escherichia coli against multiple phages and is specifically activated by structurally related, phage-encoded helicases, including UvsW and D10 from phages T4 and T5. We show that these helicases trigger effector-mediated toxicity, leading to abortive infection, which is associated with phosphoribosyl pyrophosphate depletion due to PRTase activation, thereby altering nucleotide metabolism. UvsW interacts directly with multicopy single-stranded DNA, and effector protein activation requires the catalytic activity of phage-encoded helicases. Our data indicate that activation occurs without detectable effector release or measurable changes in the composition of the Retron complex. Beyond its defensive role, Retron-Eco11 enables targeted genome editing in bacterial and eukaryotic cells, underscoring the biological relevance and biotechnological potential of type III-A3 retrons.
|
|
Scooped by
mhryu@live.com
December 28, 12:45 PM
|
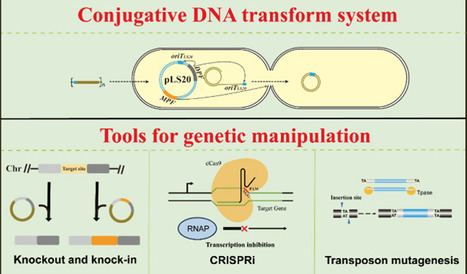
Bacillus licheniformis is a spore-forming bacterium with probiotic, environmental, and industrial applications. Many wild strains with diverse functions have been described in recent years. Nevertheless, the lack of efficient and universal genetic manipulation tools hinders the study and engineering of these strains. Here, a versatile and simple genetic manipulation toolkit is established for B. licheniformis. The cornerstone of this toolkit is a conjugative DNA transfer system. This system could effectively transfer temperature-sensitive plasmid pTSMK into all ten tested B. licheniformis strains, with efficiencies ranging from 10–5 to 10–3. Based on this DNA transfer system, the tools for maker-free knockout and knock-in, CRISPRi, as well as transposon mutagenesis, were built. A transposition frequency of 7.68 × 10–3 was observed. The toolkit developed in this study fulfills most tasks in the engineering of this species and will promote the basic and applied research of B. licheniformis.
|




























automation