 Your new post is loading...

|
Scooped by
mhryu@live.com
April 10, 5:01 PM
|
Biological nitrogen fixation in symbiotic diazotrophs is subject to oxygen regulation by an oxygen-sensing FixLJ two-component system under micro-oxic conditions. However, it remains unclear whether this mechanism is conserved in free-living diazotrophs. In this study, we discovered for the first time that FixLJ strongly inhibits the expression of nifHDK genes that encode molybdenum nitrogenase in response to oxygen. The deletion of fixLJ genes, whose expression was stimulated by oxygen, allowed a free-living photosynthetic diazotroph Rhodopseudomonas palustris to express active nitrogenase and grow diazotrophically even under oxic conditions. The unphosphorylated FixJ protein showed high-affinity binding to the promoter of nitrogenase gene cluster (PnifH) and strongly repressed the nitrogenase expression in response to oxygen. The transcriptional repression of nifHDK by FixJ reveals a new regulatory role for the FixLJ system. In addition, transcriptome analysis suggested that the FixLJ regulatory system also plays a role in the energy metabolism of R. palustris, probably through FixK regulation. This newly identified mechanism is speculated to allow R. palustris to rapidly shut down the synthesis of nitrogenase when exposed to oxygen, avoiding the build-up of nitrogenase with impaired activity due to the lack of protection from oxygen damage.
|
|
Scooped by
mhryu@live.com
April 10, 2:58 PM
|
Fusarium root rot, predominantly caused by Fusarium falciforme, poses a significant threat to soybean productivity globally. Microbiome-based strategies offer sustainable alternatives, but the mechanisms underlying multi-niche interactions remain elusive. Here, we found that a tolerant soybean cultivar (GXD2) coordinates spatially resolved metabolite signals to recruit beneficial microbes across the rhizosphere, root endosphere, and leaf endosphere. Specifically, formononetin and maltol selectively enrich Bacillus and Massilia in the rhizosphere; arctigenin and isovanillic acid recruit Bacillus and Streptomyces to the root endosphere; and flavonoids such as diosmetin attract Penicillium and Aspergillus to the leaf endosphere. Leveraging these interactions, we constructed different types of synthetic communities (SynComs) via top-down (host-selected taxa) and bottom-up (antagonist-based) strategies. Both SynComs suppressed root rot in susceptible cultivars, with foliar application of top-down SynComs significantly enhancing shoot growth. Transcriptomics revealed distinct modes of actions, that top-down SynComs activated mitogen-activated protein kinase (MAPK)-linked terpenoid and flavonoid pathways, whereas bottom-up SynComs primarily modulated host carbon–nitrogen allocation, effectively limiting pathogen resources. Our findings unveil a "metabolite-mediated, multi-niche collaborative defense" model, presenting a robust framework for microbiome-based disease management and paving the way toward sustainable crop protection strategies.
|
|
Scooped by
mhryu@live.com
April 10, 2:37 PM
|
Asgard archaea hold a pivotal position in the tree of life as the closest known relatives to eukaryotes and are therefore crucial for understanding eukaryogenesis. Earlier genomic analyses revealed that Asgard genomes are remarkably larger than those of other archaea and contain a significant number of genes seemingly acquired from bacteria. However, the precise contributions of horizontal gene transfer and gene duplication in shaping Asgard genomes remain largely unknown. Here, we present a comprehensive phylogenomic analysis to dissect the evolutionary dynamics of Asgard genomes, quantifying gene duplication, loss, and both inter- and intra-domain gene transfer events. Our findings reveal that gene transfer is widespread throughout Asgard evolution, predominantly affecting metabolic genes at the periphery of interaction networks. However, our analyses demonstrate that gene duplications, rather than horizontal gene transfers, are the primary drivers behind the increased genome sizes observed in Asgard archaea. This unique evolutionary signature in Asgard archaea—a blend of pervasive prokaryotic-like gene transfer alongside significant eukaryotic-like gene duplication—is consistent with their phylogenetic placement and offers novel insights into the genomic transitions that likely underpinned eukaryogenesis.
|
|
Scooped by
mhryu@live.com
April 10, 2:10 PM
|
Microbes frequently encounter fluctuating environments, requiring dynamic energy management strategies for survival. While carbon storage polymers like polyhydroxybutyrate (PHB) are ubiquitous across bacterial taxa, their precise ecological advantage remains poorly understood (1). Here we show that carbon storage drives conditional fitness during environmental transitions. Using a high-throughput single-cell microfluidic platform, we tracked tens of thousands of Cupriavidus necator cells under precisely controlled carbon and nitrogen fluctuations. We found that PHB provides no advantage under nutrient abundance but becomes decisive at starvation boundaries: during carbon starvation, it enables ~30% more progeny before arrest; during recovery from nitrogen starvation, it shortens lag and accelerates regrowth. Strikingly, at the single-cell level, PHB granules are inherited in an asymmetric, all-or-nothing fashion, concentrating resources into specific lineages to overcome the discrete energetic threshold required for cell division. Despite this single-cell variance, at the population level, PHB fractions robustly return to a common setpoint after nutrient shifts - a homeostatic behavior consistent with integral feedback control. These findings reveal that while PHB does not increase the basal exponential growth rate, it confers a distinct fitness advantage by prolonging the proliferative phase during nutrient depletion and facilitating successful recovery from starvation, explaining the evolutionary persistence of carbon storage in environments with pulsed resource availability.
|
|
Scooped by
mhryu@live.com
April 10, 1:57 PM
|
Small secreted peptides (SSPs) are essential signaling molecules in plants, yet their deep evolutionary origins remained unclear. Here, we integrated sensitive homology searches with rigorous filtering to systematically identify SSP and leucine-rich repeats receptor-like kinase (LRR-RLK) homologs across the EukProt database. We recovered 59 high-confidence SSP homologs from diverse deeply branching eukaryotic supergroups, with protistan sequences consistently placed at the base of phylogenies. Parallel searches recovered 916 LRR-RLKs from non-vascular plant lineages, whereas the SSP-associated XI subfamily was largely confined to Archaeplastida. Co-occurrence of SSPs and LRR-RLKs in 6 lineages, together with bidirectional motif traceability revealing strong purifying selection on core residues, supports a model in which core components of SSP signaling emerged early in eukaryotic evolution and were vertically inherited, with subsequent lineage-specific diversification and co-option during streptophyte evolution. Our study provides a revised evolutionary framework for plant peptide signaling and a broadly applicable strategy for tracing ancient origins of lineage-specific innovations.
|
|
Scooped by
mhryu@live.com
April 10, 12:53 PM
|
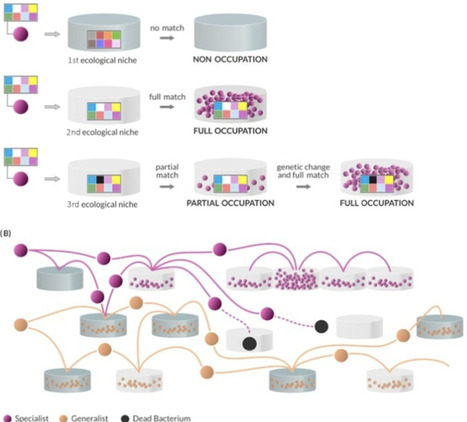
We present an alternative or complementary hypothesis to the step-by-step bacterial adaptation to specific niches. Bacterial cells spread into different environments and, by chance, encounter a suitable niche that permits reproduction. Then, they can improve their adaptation and transmit to neighboring related niches, resulting in ecogenomic variation.
|
|
Scooped by
mhryu@live.com
April 10, 1:27 AM
|
Alternative splicing (AS) of precursor mRNAs (pre-mRNAs) constitutes a major means to increase transcriptome complexity in higher eukaryotes and critically contributes to the re-programming of gene expression in response to internal and environmental signals. Technological advances have enabled us to determine transcriptome-wide AS patterns at unprecedented accuracy, depth, and throughput. Furthermore, powerful tools for examining the regulatory mechanisms underlying AS decisions have been successfully established for plants, including methods for profiling the in vivo interaction landscape of splicing regulatory proteins with their target pre-mRNAs. Combining these novel approaches with functional studies of individual AS events identified many critical components of the plant splicing code, consisting of cis-regulatory elements in the pre-mRNA and trans-acting factors, such as splicing regulatory proteins. Their concerted action affects splice site selection by the spliceosome, thereby generating highly dynamic and complex AS outputs. Here, we will review our current knowledge of AS regulation by RNA sequence and structural motifs in cis and networks of trans-acting splicing regulators. We will also discuss how, despite overall low complexity of the target motifs for binding of splicing regulators and their often-redundant functions, high levels of precision and specificity in AS can be achieved.
|
|
Scooped by
mhryu@live.com
April 10, 1:18 AM
|
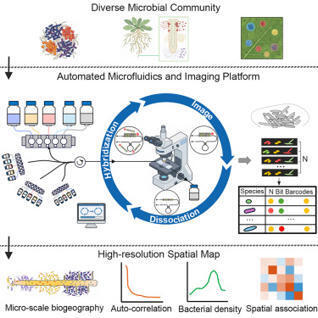
Spatial mapping of microbial communities at single-cell resolution is opening up dimensions to understand microbiome organization and function. However, current fluorescence in situ hybridization (FISH) methods for microbiomes are limited by multiplexity and scalability. Here, we present the sequential error-robust FISH spatial mapping platform (SEER-Map) for fully automated imaging of complex microbial communities. We show that an integrated platform of fluidics control and fluorescence microscopy can perform 40 rounds of sequential FISH. We apply SEER-Map to profile complex microbial communities colonized on plant roots and identify distinct spatial patterns and species co-occurrence at the micron-scale. Our work establishes SEER-Map as a high-throughput and scalable platform for high-resolution spatial profiling of microbiomes.
|
|
Scooped by
mhryu@live.com
April 10, 12:54 AM
|
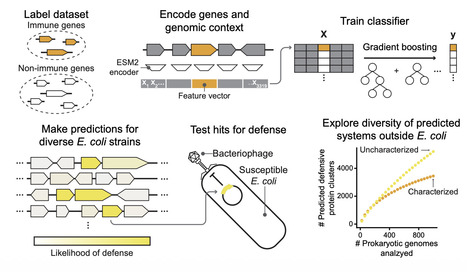
Antiphage defense systems protect bacteria from viral infection and have inspired important biotechnologies such as CRISPR-Cas9 while also revealing the evolutionary roots of eukaryotic innate immunity. Many systems have been discovered by genomic colocalization, but this approach cannot identify systems outside of defense islands. We present DefensePredictor, a machine learning model that uses protein language model embeddings to classify proteins as defensive. Applying DefensePredictor to 69 diverse E. coli strains, we predicted hundreds of previously unknown systems and experimentally validated 42 of them. Analysis of 1000 diverse prokaryotic genomes identified nearly 3000 protein clusters lacking homology to known systems, revealing a vast, uncharacterized defense repertoire. DefensePredictor will facilitate the comprehensive discovery of antiphage defense systems, which promises to reveal additional connections between prokaryotic and eukaryotic immunity and accelerate biotechnology development.
|
|
Scooped by
mhryu@live.com
April 9, 11:33 PM
|
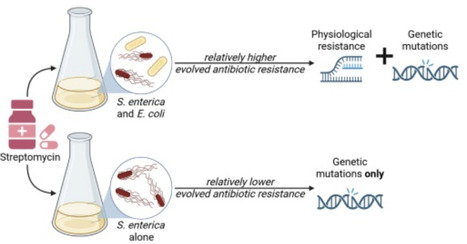
Interspecies interactions can influence the physiology of competing species, shaping their long-term evolutionary trajectories. Although interspecific competition’s role in community dynamics is well-documented, its impact on evolutionary outcomes and mechanisms is less explored. Here, we investigate how interspecies competition affects antibiotic resistance evolution in the gut pathogen Salmonella enterica within synthetic microbial communities. Specifically, we examine how the presence of an interspecific competitor, E. coli, modulates resistance evolution at low streptomycin concentrations. Our findings reveal that interspecies competition results in the selection of S. enterica mutants with higher resistance levels by increasing the likelihood of accumulating resistance mutations that follow a trajectory of negative fitness epistasis. We show that this effect is driven by the enhanced expression of the cryptic aminoglycoside transferase gene (aadA). Our study thus links antibiotic resistance evolution to competition-induced physiological changes, emphasizing the interplay between interspecies interaction and adaptation to environmental conditions.
|
|
Scooped by
mhryu@live.com
April 9, 11:26 PM
|
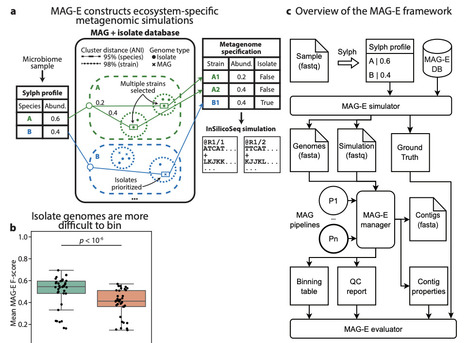
The generation of Metagenome Assembled Genomes (MAG) has become a standard and basic step in the analysis of metagenomic data. This multi-step process, which includes assembly, binning, refinement, and quality control, has many alternative approaches, algorithms, and parameters. Determining the ideal approach for a given ecosystem and study, or highlighting algorithmic gaps in need of additional research and development, requires rigorous benchmarking. We present MAG-E (MAG pipeline Evaluator), a generalizable and expandable framework for end-to-end evaluation of entire MAG pipelines: from assembly, through binning, to quality control and filtering. MAG-E relies on simulations that are built to match an ecosystem of interest and provide a ground truth for accurate evaluation. To demonstrate the capabilities of MAG-E, we benchmark two assemblers, six binning algorithms, three binning modes, and three quality control and refinement methods in the context of the human gut microbiome. Our findings offer multiple insights into optimal MAG generation in this context. We find that metaSPAdes consistently outperforms MEGAHIT in terms of recall (completeness), and that COMEBin overall outperforms alternative binning algorithms, but has lower precision than SemiBin2. While multi-sample binning results in higher precision, as previously shown, single-sample binning has higher recall and leads to better overall performance with modern binners. Binning refinement, which combines bins from multiple different algorithms, leads to reduced performance. We further show that CheckM2 systematically overestimates completeness and underestimates contamination, and that this is partially ameliorated when using GUNC. Finally, we analyze performance at the contig level, and demonstrate that binning algorithms systematically underperform for prophages and fail to bin contigs that are shared between genomes. Overall, MAG-E offers deep insights into successes and gaps in this important analytic process.
|
|
Scooped by
mhryu@live.com
April 9, 11:19 PM
|
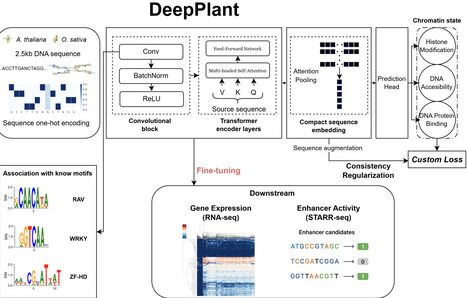
Large-scale sequence-to-function deep learning models have demonstrated unparalleled ability to model biological sequences and have revolutionized the field of regulatory genomics. However, the majority of such efforts have centered on human and mammalian systems, leaving plant regulatory genomics comparatively underexplored. To address this gap, we introduce Deep-Plant, a supervised foundation model trained to predict chromatin state directly from genomic sequence. In contrast to large language models, which are trained in a self-supervised manner using sequence alone, our model is trained to predict chromatin state across tissues and conditions. Training the model on a large collection of genome-wide experiments including DNA accessibility, transcription factor binding, and histone modifications, provides it with added biological context beyond the sequence itself. We demonstrate that the resulting model is an effective platform for developing accurate models of regulatory activity relevant to gene expression and active enhancers, exhibiting large improvements in speed, accuracy, and interpretability over the complementary approach of fine-tuning DNA language models. Deep-Plant models are available in Arabidopsis and rice, and work well as a building block for sequence modeling in related species such as corn. Together, these results establish supervised, chromatin-informed foundation models as a practical and effective paradigm for regulatory sequence modeling in plants.
|
|
Scooped by
mhryu@live.com
April 9, 10:57 PM
|
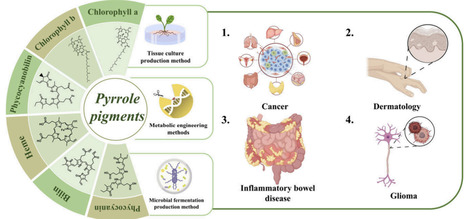
Natural food pigments primarily originate from two sources: chemical synthesis and plant-derived production. With the rapid advancement of society and technology, there is a growing demand for environmentally friendly and healthy food options. Consequently, the demand for safe, nontoxic, and sustainable sources of natural pigments has risen sharply. Natural pigments are biosynthesized during the growth and metabolic processes of plant tissues, and compounds derived from these pigments exhibit a wide range of biological activities that are beneficial to human health and disease treatment. However, due to their inherent instability and low abundance, increasing research efforts have been directed toward the bioengineering of natural pigment production. This review classifies natural pigments into five major structural categories: pyrrole, isoprenoids, quinones, phenols, and betalains. Unlike previous reviews that focused on a single pigment component or specific application fields, this review systematically integrates the biosynthetic pathways, synthetic biology strategies, pharmacological activity mechanisms, and application progress in medicine, health care, and cosmetics of natural pigment-containing medicinal materials. It emphasizes their multiple potentials as “functional pigments” in the development of natural medicines. Additionally, the review combines emerging technologies such as metabolic engineering, artificial intelligence (AI)-assisted screening, and biosensing, proposing a cross-disciplinary development path from basic synthesis to high-value applications and demonstrating strong systematicity and a forward-looking nature. It provides a new integrated perspective for innovative research on natural pigment components.
|
|
|
Scooped by
mhryu@live.com
April 10, 3:00 PM
|
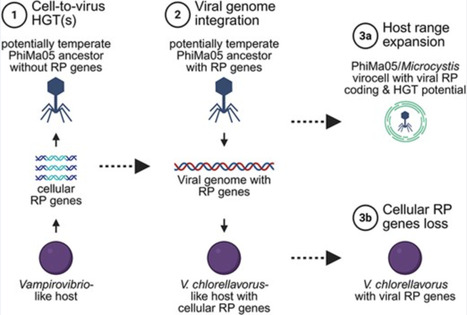
It has been proposed that a defining distinction between viruses and cells lies in the absence or presence of ribosomal genes, respectively. Recent studies revealing that viruses occasionally encode ribosomal proteins (RPs) have challenged this view. However, so far, only viral genomes with up to three RPs have been discovered. Here, we perform a functional genome analysis of the Microcystis jumbo phage PhiMa05 and show that it encodes six RPs, an RP acetyltransferase, and a ribosome biogenesis protein. To our knowledge, this makes PhiMa05 the first cyanophage reported to encode RPs, as well as the virus with the most comprehensive RP-coding set of the known virosphere. Evolutionary analyses suggest that these viral RP-coding genes may have been horizontally transferred from a temperate ancestor of PhiMa05 to certain members of the Vampirovibrionia, a non-photosynthetic basal lineage of Cyanobacteriota, via the integration of the viral genome. We find that four RPs, the RP acetyltransferase, and the ribosome biogenesis protein of the PhiMa05-like prophages are the only copies of those proteins that the near-complete genomes of some Vampirovibrio hosts possess. We hypothesize that such cellular organisms may depend on the PhiMa05-like prophage for protein synthesis, and hence life itself. Collectively, our results provide evidence for the existence of viruses with particularly enriched sets of RP-coding genes and indicate that, in some cases, such viral genes have been transferred to cells, potentially becoming essential for the survival of the host.
|
|
Scooped by
mhryu@live.com
April 10, 2:45 PM
|
Investigating the lethal effect of multi-gene knockout is essential for discovering novel antibiotic targets and metabolic engineering. Unlike single genes or gene pairs, three-gene combinations involve more intricate interactions, making experimental screening time-consuming. Computational methods, particularly Genome-scale metabolic model (GEM)-based Flux balance analysis (FBA), requires constructing new GEMs from experimental data, limiting its use for new species. Moreover, using FBA for three-gene knockout screening could take several years. Therefore, a faster and GEM-independent approach is needed to facilitate genome-wide three-gene knockout screening. Here, we introduce Tripleknock for predicting the lethal effects of three-gene knockouts. Tripleknock was trained using genome-derived protein sequence features from E. coli K-12 MG1655, and three-gene knockout simulations using FBA. The model uses a threshold of 90% reduction in cell growth to define lethal effect as the prediction output. Compared to FBA, Tripleknock yields predictions ~ 20 × faster and achieves an average cross-species F1 score of 0.77 on six Enterobacteriaceae pathogens (FBA-defined labels). To mitigate information leakage, we additionally evaluated Tripleknock under a strict gene-level disjoint Train/Val/Test split (genes left out). We further benchmarked against an essentiality-rule baseline and literature-curated (n = 37) E. coli triple perturbations for external validation; no false positives were observed among predicted lethal cases (FP = 0) in this small curated set. To the best of our knowledge, this provides a reproducible baseline and evaluation framework for bacterial triple-knockout lethality prediction.
|
|
Scooped by
mhryu@live.com
April 10, 2:21 PM
|
Photosynthetic cyanobacteria are promising platforms for sustainable chemical production, as they can convert light and CO₂ into valuable compounds. Achieving this often requires engineering cyanobacteria with non-native enzymes with strong promoters to maximize enzyme accumulation. However, despite extensive engineering efforts, the extent to which heterologous proteins misfold and undergo degradation in cyanobacteria remains unknown. Here, we systematically investigate the fate of recombinant proteins in Synechocystis sp. PCC 6803 by quantifying metabolic enzyme degradation. To do this, we developed a quantitative approach that combines split-GFP protein reporting with inducible CRISPRi knockdown of Clp protease system, enabling detection of proteins that would otherwise be degraded. Applying this method to 103 heterologous proteins previously used in cyanobacterial metabolic engineering studies, we find that nearly half undergo significant degradation, with some losing over 95% of their potential expression. Furthermore, we demonstrate that replacing enzymes with homologs is often a more effective strategy to address expression issues than optimizing genetic elements. These findings provide the first quantitative overview of heterologous protein expression in cyanobacteria and identify enzymes that are poorly expressed and suboptimal for their respective pathways, information usable to increase production titers in photosynthetic cell factories.
|
|
Scooped by
mhryu@live.com
April 10, 2:01 PM
|
De novo gene emergence from non-coding sequences is increasingly recognized as an important evolutionary mechanism, yet the functional potential of random sequences remains debated. Previous experiments suggested that expression of random sequence clones in E. coli can enhance growth of the cells bearing them, i.e. they provide a fitness advantage. However, these findings have been questioned, regarding potential confounding effects of the clone mixtures and a possibly negatively acting peptide expressed from the cloning vector. Here we performed controlled competitive growth assays using a defined subset of 64 random sequence clones representing a spectrum of fitness effects. Experiments across multiple conditions, including two different growth cycle durations, induction states, and replicate sets, showed high technical reproducibility and consistent clone-specific growth trajectories for the majority of the clones, but for some also influences of genomic background and experimental conditions. While vector-derived constructs that inhibit the vector-coded peptide expression showed the same fitness improvements relative to the parental vector that were previously shown, several random sequence clones exhibited higher positive selection coefficients under conditions of exponential growth. These effects persisted even when negative clones were excluded, indicating that they are not driven by competition dynamics with negative clones. Our results demonstrate that positive growth effects of random sequence clones cannot be explained by clone mixture and vector artifacts alone. Instead, a subset of random sequences confers genuine fitness advantages comparable to beneficial mutations observed in experimental evolution studies. These findings provide strong experimental support for the capacity of random sequences to generate adaptive functions and underscore their role in de novo gene evolution.
|
|
Scooped by
mhryu@live.com
April 10, 1:45 PM
|
Light is one of the most pervasive physical cues in aquatic environments, yet its impact on nonphototrophic pathogens remains largely unexplored. Here, we show that a strain of cholera bacterium Vibrio cholerae directly couples illumination to motility through cyclic AMP (cAMP) signaling. Exposure to visible light rapidly elevates intracellular cAMP and increases swimming speed, whereas deletion of the single adenylyl cyclase gene (cyaA) abolishes both responses; complementation or addition of exogenous cAMP restores the phenotype. Heterologous expression of V. cholerae CyaA in an E. coli ΔcyaA ΔcpdA background reconstitutes light-activated cAMP synthesis, indicating that CyaA confers photoreactivity. Purified CyaA exhibits a reversible light-dependent spectral shift consistent with flavin-dependent photochemistry, identifying it as a light-responsive cyclase. Illumination triggers rapid membrane hyperpolarization and sodium efflux, strengthening the sodium-motive force that powers the flagellar motor. This response persists under nutrient-limited conditions. Together, these findings define a light → cAMP → sodium-motive force coupling axis in V. cholerae, suggesting that ambient light may influence motility and dispersal in sunlit environments.
|
|
Scooped by
mhryu@live.com
April 10, 10:00 AM
|
CRISPR/Cas13a is a powerful RNA-targeting platform for molecular diagnostics, but conventional single-effector systems typically require contiguous RNA targets longer than ∼20–28 nt, limiting sensitivity and target flexibility. CRISPR/Cas13a-CTAM is presented as a compensatory target activation mechanism that facilitates synergistic Cas13a activation through two independently programmable short RNA effectors. By functionally decoupling allosteric activation and binding stabilization, CRISPR/Cas13a-CTAM supports robust activation by ultra-short RNA targets as short as 13 nt, substantially expanding the detectable target range. Compared with traditional single-effector Cas13a assays, CRISPR/Cas13a-CTAM achieves a detection limit of 1 fM for a 13-nt RNA target, representing an approximately tenfold sensitivity improvement. Notably, a single-nucleotide mismatch within the 13-nt target induces up to a 35-fold reduction in apparent cleavage rate, corresponding to a sevenfold enhancement in mismatch discrimination. The dual-effector architecture further enables simultaneous dual-target detection, demonstrated by dual miRNA profiling related to COVID-19 and combined detection of exosome membrane proteins. Moreover, the weakly activating effector was utilized as an anchoring module to achieve the first functional immobilization of Cas13a on a sensing surface, enabling in situ electrochemical miRNA detection. By overcoming the reliance on long RNA targets, CRISPR/Cas13a-CTAM provides a sensitive, programmable platform for RNA diagnostics and integrated biosensor development.
|
|
Scooped by
mhryu@live.com
April 10, 1:23 AM
|
Alternative splicing (AS) represents a pivotal post-transcriptional regulatory mechanism, profoundly expanding proteomic diversity and functional complexity by enabling single genes to generate multiple mRNA isoforms. In plants, AS serves as a survival toolkit, dynamically modulating stress-responsive signaling pathways, transcriptional networks, and protein functional specialization to optimize environmental fitness. Recent advances in high-throughput sequencing technologies and computational tools have significantly deepened our understanding of AS regulation in plants. Notably, breakthroughs such as long-read transcriptome sequencing and single-cell RNA analysis have revolutionized the resolution at which we can characterize AS landscapes. These developments have collectively illuminated the critical role of AS in mediating plant responses to diverse abiotic stresses, including drought, salinity, and extreme temperatures. The resulting discoveries have opened transformative avenues for crop improvement through precise manipulation of splicing patterns. Innovative strategies such as CRISPR-Cas9-based splice editing and engineered splicing factors now provide powerful platforms for developing climate-resilient, high-yielding crop varieties with enhanced stress tolerance and nutritional quality. Here, we systematically examine the molecular mechanisms underlying AS-mediated plant stress responses, and cutting-edge applications of AS engineering in precision agriculture. By synthesizing fundamental insights with biotechnological innovations, we highlight the transformative potential of AS manipulation in addressing the pressing global agricultural challenges.
|
|
Scooped by
mhryu@live.com
April 10, 1:12 AM
|
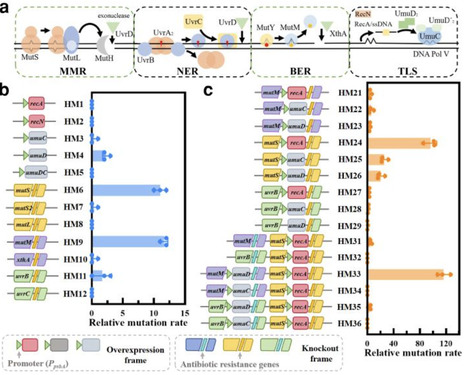
Cyanobacteria represent an ancient group of photosynthetic microorganisms that offer unparalleled insights into evolutionarily conserved stress adaptation mechanisms essential for plant resilience. To investigate how photosynthetic organisms mitigate chemical stressors, we employed Synechocystis sp. PCC 6803—a keystone model for photosynthetic research due to its plant-like electron transport chain and stress-responsive plasticity. By implementing a genomic hypermutation strategy, we synergistically knocked out DNA replication fidelity genes and overexpressed error-prone replication elements, generating hypermutable strains HM24 and HM33 with relative mutation rates of 97 and 116-fold, respectively. Following triclosan (TCS) stress screening, the CRISPR-Cpf1 strategy was used to complement mutations and yielded transformants R-HM24 and R-HM33 that exhibited 96 h EC50 values of 4.963 and 5.238 mg/L representing 322- and 340-fold increases over wild-type levels, respectively. The strains demonstrated enhanced TCS and multidrug antibiotic tolerance. Whole-genome resequencing identified consistent missense mutation in fabI across resistant strains. Mechanistic analyses revealed that the hypermutated Synechocystis strains acquired resistance primarily by mutating the essential fabI protein to decrease its affinity for TCS. This study establishes the application of hypermutation-driven evolution for rapid dissection of pollutant resistance in photosynthetic microbes, thereby advocating for stricter regulation of antimicrobial pollutants in aquatic environments.
|
|
Scooped by
mhryu@live.com
April 10, 12:50 AM
|
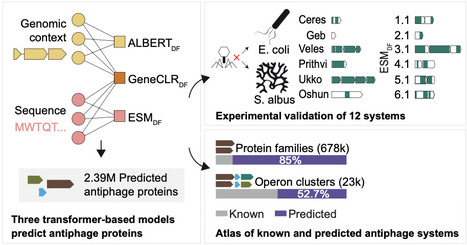
The bacterial pangenome contains a vast diversity of antiphage systems, whose overall extent is still unknown. In this study, we developed complementary machine learning approaches to systematically predict antiphage function from genomic context, protein sequence, or their combination, achieving up to 99% precision and 92% recall. We validated these models experimentally in Escherichia and Streptomyces with the discovery of 12 antiphage systems. Applied to over 32,000 bacterial genomes, these models expand the predicted antiphage repertoire, with ~1.5% of bacterial genomes devoted to defense and more than 85% of predicted protein families remaining uncharacterized. We provide an interactive catalog of more than 19,000 candidate operon families for experimental follow-up. Together, these findings show that most molecular diversity in bacterial immunity remains uncharacterized and provide a foundation for its systematic exploration.
|
|
Scooped by
mhryu@live.com
April 9, 11:30 PM
|
Scientific breakthroughs infrequently translate into startups without structured training. Stanford’s BioEntrepreneurship Bootcamp teaches a practical framework for navigating bench-to-business translation. The curriculum integrates instruction on intellectual property, team formation, risk assessments, milestone-based funding, and investor communication, with the goal of equipping scientists to advance discoveries into viable ventures.
|
|
Scooped by
mhryu@live.com
April 9, 11:22 PM
|
Generating graphical diagrams of microbial and organellar genomes is a common and essential task in bioinformatics. Existing tools often present a trade-off; while powerful programming libraries that require coding skills, graphical applications require server processing or local installation with complex dependency. This highlights the need for a tool that offers both programmatic control for batch processing and graphical accessibility for ease of use. To fill this gap, I developed gbdraw, a web application that generates circular and linear genome diagrams from self-contained GenBank or DDBJ files or combinations of GFF3 annotation and FASTA sequence files. Its core functions include visualizing annotated features, plotting GC content/skew tracks, and optionally generating pairwise sequence comparisons for comparative genomics. It is available as both a GUI web application and a command-line utility. Unlike existing web-based tools that require data upload to a remote server, gbdraw operates entirely within the user's web browser. This serverless architecture ensures that sensitive sequence data never leaves the local machine, providing a secure environment for visualizing unpublished genomic data. Availability and Implementation: gbdraw is implemented in Python 3 (version 3.10+) and is freely available under the MIT license. The web app is available at https://gbdraw.app/. Source code and documentation are available at https://github.com/satoshikawato/gbdraw. The local version can be installed from the Bioconda channel using a conda-compatible package manager.
|
|
Scooped by
mhryu@live.com
April 9, 11:00 PM
|
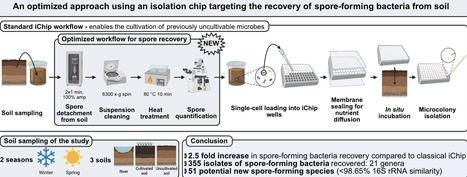
Spore-forming bacteria are key candidates for applications in biotechnology and biocontrol due to their resistance to physical and chemical stress and metabolic diversity. However, their recovery from soil is often hindered by limitations in conventional cultivation methods. The isolation chip (iChip) has expanded access to previously uncultivable microorganisms. This study aimed to optimize pretreatment and incubation conditions for the selective isolation of spore-forming bacteria using a modified iChip protocol. Soil samples were collected from three environments in Brittany, France, during two seasons. Pretreatment included sequential washing, sonication, centrifugation, and heat treatment at 80 °C to select spores while minimising soil particules to allow accurate microscopic quantification. Stainless steel iChip plates with 96 wells were designed to improve membrane sealing and handling. Plates were inoculated with spore-enriched suspensions and incubated in situ for up to six weeks before recovery at the laboratory at two temperatures (15 °C and 30 °C).A total of 757 isolates were obtained, with 52% identified as spore-formers, a substantial increase compared with unmodified iChip workflows (~21%). Taxonomic analysis revealed 79 genera, including Bacillus, Paenibacillus. Phylogenetic analysis highlighted 51 isolates with less than 98.65% 16S rRNA similarity to known species, suggesting potential novelty. The recovery of spore-formers can be achieved through selective pretreatments. Even rare taxa can be revealed with this in situ cultivation. The optimized method offers a promising framework for the recovery of uncultured spore-forming bacteria with potential applications in biocontrol, biotechnology, and natural product discovery.
|



















lee sy