Your new post is loading...
Your new post is loading...

|
Rescooped by
Kamoun Lab @ TSL
from Plants and Microbes
February 14, 2017 6:17 AM
|
Background. The prevailing paradigm of host-parasite evolution is that arms races lead to increasing specialisation via genetic adaptation. Insect herbivores are no exception and the majority have evolved to colonise a small number of closely related host species. Remarkably, the green peach aphid, Myzus persicae, colonises plant species across 40 families and single M. persicae clonal lineages can colonise distantly related plants. This remarkable ability makes M. persicae a highly destructive pest of many important crop species.
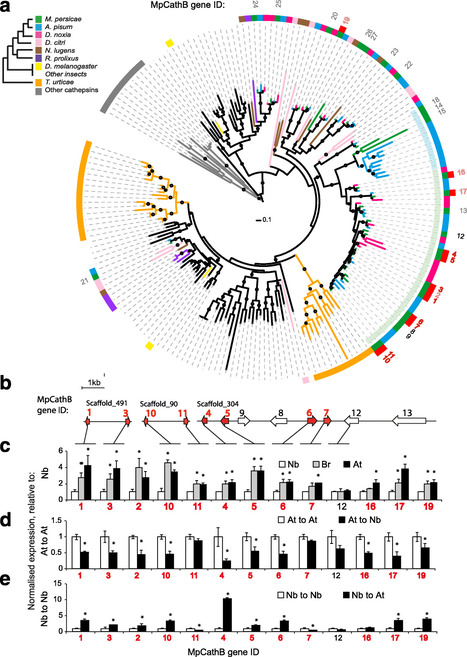
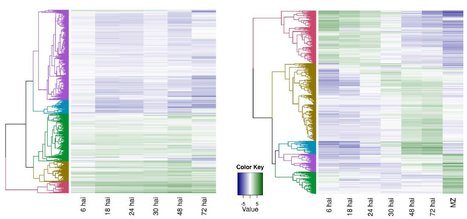
Results. To investigate the exceptional phenotypic plasticity of M. persicae, we sequenced the M. persicae genome and assessed how one clonal lineage responds to host plant species of different families. We show that genetically identical individuals are able to colonise distantly related host species through the differential regulation of genes belonging to aphid-expanded gene families. Multigene clusters collectively upregulate in single aphids within two days upon host switch. Furthermore, we demonstrate the functional significance of this rapid transcriptional change using RNA interference (RNAi)-mediated knock-down of genes belonging to the cathepsin B gene family. Knock-down of cathepsin B genes reduced aphid fitness, but only on the host that induced upregulation of these genes.
Conclusions. Previous research has focused on the role of genetic adaptation of parasites to their hosts. Here we show that the generalist aphid pest M. persicae is able to colonise diverse host plant species in the absence of genetic specialisation. This is achieved through rapid transcriptional plasticity of genes that have duplicated during aphid evolution.
|
|
Scooped by
Kamoun Lab @ TSL
January 30, 2017 6:28 AM
|
Background. Plant-pathogenic oomycetes are responsible for economically important losses on crops worldwide. Phytophthora palmivora, a broad-host-range tropical relative of the potato late blight pathogen, causes rotting diseases in many important tropical crops including papaya, cocoa, oil palm, black pepper, rubber, coconut, durian, mango, cassava and citrus. Transcriptomics have helped to identify repertoires of host-translocated microbial effector proteins which counteract defenses and reprogram the host in support of infection. As such, these studies have helped understanding of how pathogens cause diseases. Despite the importance of P. palmivora diseases, genetic resources to allow for disease resistance breeding and identification of microbial effectors are scarce. Results. We employed the model plant Nicotiana benthamiana to study the P. palmivora root infections at the cellular and molecular level. Time-resolved dual transcriptomics revealed different pathogen and host transcriptome dynamics. De novo assembly of P. palmivora transcriptome and semi-automated prediction and annotation of the secretome enabled robust identification of conserved infection-promoting effectors. We show that one of them, REX3, suppresses plant secretion processes. In a survey for early transcriptionally activated plant genes we identified a N. benthamiana gene specifically induced at infected root tips that encodes a peptide with danger-associated molecular features. Conclusions. These results constitute a major advance in our understanding of P. palmivora diseases and establish extensive resources for P. palmivora pathogenomics, effector-aided resistance breeding and the generation of induced resistance to Phytophthora root infections. Furthermore, our approach to find infection relevant secreted genes is transferable to other pathogen-host interactions and not restricted to plants.
|
|
Scooped by
Kamoun Lab @ TSL
December 15, 2016 4:55 AM
|
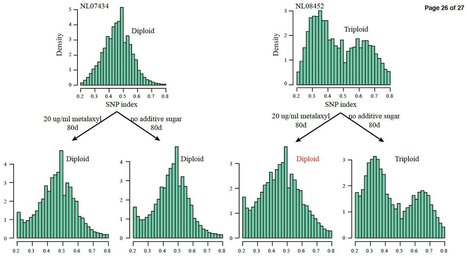
The oomycete Phytophthora infestans was the causal agent of the Irish Great Famine and is a recurring threat to global food security. The pathogen can reproduce both sexually and asexually, with high potential to adapt to various environments and great risk to break disease resistance genes in potato. As other oomycetes, P. infestans is regarded to be diploid during the vegetative phase of its life cycle, although some studies reported trisomy, and polyploidy. Using microsatellite fingerprinting, genome-wide assessment of SNP polymorphism, nuclear DNA quantification, and microscopic counting of chromosome numbers we assessed the ploidy level of isolates. All progeny from sexual populations of P. infestans in nature were found to be diploid, in contrast nearly all dominant asexual lineages, including the most important pandemic clonal lineages US-1 and 13_A2 were triploid. Such triploids possess significantly more allelic variation than diploids. We observed that triploid genotype can change to a diploid genome constitution when exposed to artificial stress conditions. This study reveals that fluctuations in the ploidy level maybe a key factor in the adaptation process of this notorious plant destroyer and imposes an extra challenge to control this disease.
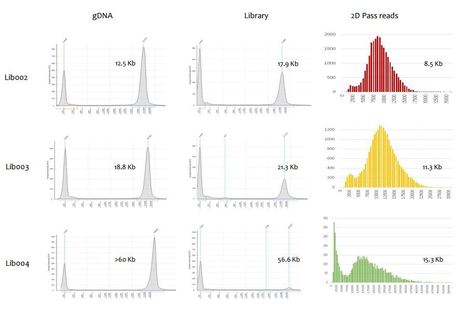
The ability to quickly obtain accurate genome sequences of eukaryotic pathogens at low costs provides a tremendous opportunity to identify novel targets for therapeutics, develop pesticides with increased target specificity and breed for resistance in food crops. Here, we present the first report of the ~54 MB eukaryotic genome sequence of Rhizoctonia solani, an important pathogenic fungal species of maize, using nanopore technology. Moreover, we show that optimizing the strategy for wet-lab procedures aimed to isolate high quality and ultra-pure high molecular weight (HMW) DNA results in increased read length distribution and thereby allowing generation of the most contiguous genome assembly for R. solani to date. We further determined sequencing accuracy and compared the assembly to short-read technologies. With the current sequencing technology and bioinformatics tool set, we are able to deliver an eukaryotic fungal genome at low cost within a week. With further improvements of the sequencing technology and increased throughput of the PromethION sequencer we aim to generate near-finished assemblies of large and repetitive plant genomes and cost-efficiently perform de novo sequencing of large collections of microbial pathogens and the microbial communities that surround our crops.
Via Steve Marek, Jessie Uehling
|
|
Rescooped by
Kamoun Lab @ TSL
from TAL effector science
September 29, 2016 12:45 PM
|
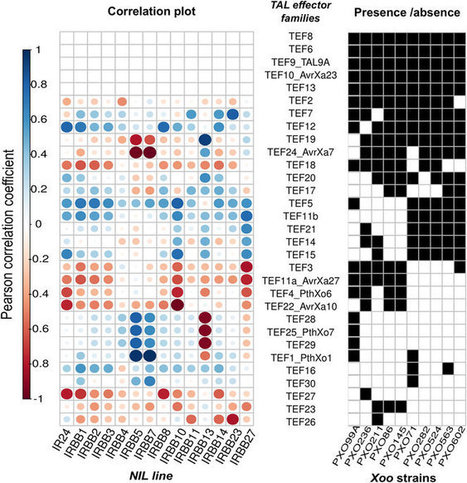
Understanding the processes that shaped contemporary pathogen populations in agricultural landscapes is quite important to define appropriate management strategies and to support crop improvement efforts. Here, we took advantage of an historical record to examine the adaptation pathway of the rice pathogen Xanthomonas oryzae pv. oryzae (Xoo) in a semi-isolated environment represented in the Philippine archipelago. By comparing genomes of key Xoo groups we showed that modern populations derived from three Asian lineages. We also showed that diversification of virulence factors occurred within each lineage, most likely driven by host adaptation, and it was essential to shape contemporary pathogen races. This finding is particularly important because it expands our understanding of pathogen adaptation to modern agriculture.
Via dromius
|
|
Rescooped by
Kamoun Lab @ TSL
from Phytophthora biology
September 8, 2016 12:28 PM
|
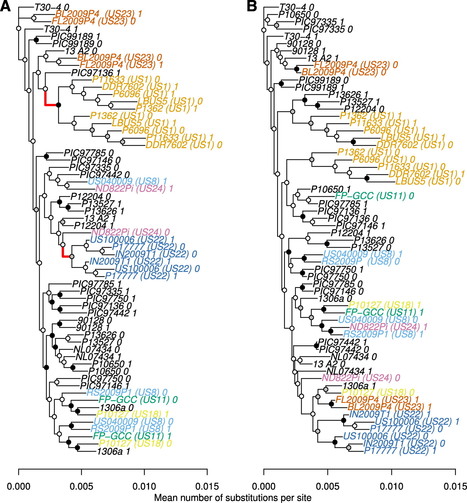
Populations of the potato and tomato late-blight pathogen Phytophthora infestans are well known for emerging as novel clonal lineages. These successions of dominant clones have historically been named US1 through US24, in order of appearance, since their first characterization using molecular markers. Hypothetically, these lineages can emerge through divergence from other U.S. lineages, recombination among lineages, or as novel, independent lineages originating outside the United States. We tested for the presence of phylogenetic relationships among U.S. lineages using a population of 31 whole-genome sequences, including dominant U.S. clonal lineages as well as available samples from global populations. We analyzed ancestry of the whole mitochondrial genome and samples of nuclear loci, including supercontigs 1.1 and 1.5 as well as several previously characterized coding regions. We found support for a shared ancestry among lineages US11 and US18 from the mitochondrial genome as well as from one nuclear haplotype on each supercontig analyzed. The other nuclear haplotype from each sample assorted independently, indicating an independent ancestry. We found no support for emergence of any other of the U.S. lineages from a common ancestor shared with the other U.S. lineages. Each of the U.S. clonal lineages fit a model where populations of new clonal lineages emerge via migration from a source population that is sexual in nature and potentially located in central Mexico or elsewhere. This work provides novel insights into patterns of emergence of clonal lineages in plant pathogen genomes.
Via Niklaus Grunwald
|
|
Scooped by
Kamoun Lab @ TSL
September 2, 2016 11:34 AM
|
Since the late 1990s, UK farmers growing barley have seen the yields and quality of their harvests hurt by an emerging disease called Ramularia leaf spot. The disease is caused by the pathogenic fungus Ramularia collo-cygni. Now a team of scientists studying this fungus have sequenced and explored its genome. Their work helps to illuminate how the fungus causes disease in barley, and enables future studies to investigate why Ramularia leaf spot has become a threat to barley production and a serious economic problem. The scientists performed the work at Scotland’s Rural College (SRUC), The University of Edinburgh's Institute of Evolutionary Biology and Edinburgh Genomics facility, and Rothamsted Research, which receives strategic funding from BBSRC. The study is published this week in the journal BMC Genomics.
|
|
Scooped by
Kamoun Lab @ TSL
June 29, 2016 11:11 AM
|
Pathogenicity islands are sets of successive genes in a genome that determine the virulence of a bacterium. In a growing number of studies, bacterial virulence appears to be determined by multiple islands scattered along the genome. This is the case in a family of seven plant pathogens and a human pathogen that, under KdgR regulation, massively secrete enzymes such as pectinases that degrade plant cell wall. Here we show that their multiple pathogenicity islands form together a coherently organized, single “archipelago” at the genome scale. Furthermore, in half of the species, most genes encoding secreted pectinases are expressed from the same DNA strand (transcriptional co-orientation). This genome architecture favors DNA conformations that are conducive to genes spatial co-localization, sometimes complemented by co-orientation. As proteins tend to be synthetized close to their encoding genes in bacteria, we propose that this architecture would favor the efficient funneling of pectinases at convergent points within the cell. The underlying functional hypothesis is that this convergent funneling of the full blend of pectinases constitutes a crucial strategy for successful degradation of the plant cell wall. Altogether, our work provides a new approach to describe and predict, at the genome scale, the full virulence complement.
|
|
Rescooped by
Kamoun Lab @ TSL
from Plant pathogenic fungi
May 28, 2016 4:01 AM
|
One major threat to global food security that requires immediate attention, is the increasing incidence of host shift and host expansion in growing number of pathogenic fungi and emergence of new pathogens. The threat is more alarming because, yield quality and quantity improvement efforts are encouraging the cultivation of uniform plants with low genetic diversity that are increasingly susceptible to emerging pathogens. However, the influence of host genome differentiation on pathogen genome differentiation and its contribution to emergence and adaptability is still obscure. Here, we compared genome sequence of 6 isolates of Magnaporthe species obtained from three different host plants. We demonstrated the evolutionary relationship between Magnaporthe species and the influence of host differentiation on pathogens. Phylogenetic analysis showed that evolution of pathogen directly corresponds with host divergence, suggesting that host-pathogen interaction has led to co-evolution. Furthermore, we identified an asymmetric selection pressure on Magnaporthe species. Oryza sativa-infecting isolates showed higher directional selection from host and subsequently tends to lower the genetic diversity in its genome. We concluded that, frequent gene loss or gain, new transposon acquisition and sequence divergence are host adaptability mechanisms for Magnaporthe species, and this coevolution processes is greatly driven by directional selection from host plants.
Via Steve Marek
|
|
Rescooped by
Kamoun Lab @ TSL
from Plants and Microbes
May 24, 2016 9:17 AM
|
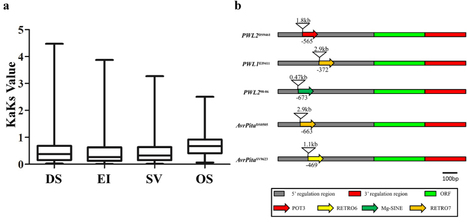
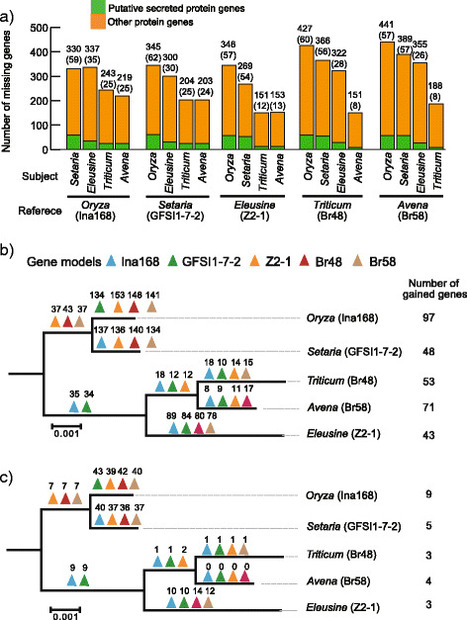
Background. Magnaporthe oryzae (anamorph Pyricularia oryzae) is the causal agent of blast disease of Poaceae crops and their wild relatives. To understand the genetic mechanisms that drive host specialization of M. oryzae, we carried out whole genome resequencing of four M. oryzae isolates from rice (Oryza sativa), one from foxtail millet (Setaria italica), three from wild foxtail millet S. viridis, and one isolate each from finger millet (Eleusine coracana), wheat (Triticum aestivum) and oat (Avena sativa), in addition to an isolate of a sister species M. grisea, that infects the wild grass Digitaria sanguinalis. Results. Whole genome sequence comparison confirmed that M. oryzae Oryza and Setaria isolates form a monophyletic and close to another monophyletic group consisting of isolates from Triticum and Avena. This supports previous phylogenetic analysis based on a small number of genes and molecular markers. When comparing the host specific subgroups, 1.2–3.5 % of genes showed presence/absence polymorphisms and 0–6.5 % showed an excess of non-synonymous substitutions. Most of these genes encoded proteins whose functional domains are present in multiple copies in each genome. Therefore, the deleterious effects of these mutations could potentially be compensated by functional redundancy. Unlike the accumulation of nonsynonymous nucleotide substitutions, gene loss appeared to be independent of divergence time. Interestingly, the loss and gain of genes in pathogens from the Oryza and Setaria infecting lineages occurred more frequently when compared to those infecting Triticum and Avena even though the genetic distance between Oryza and Setaria lineages was smaller than that between Triticum and Avena lineages. In addition, genes showing gain/loss and nucleotide polymorphisms are linked to transposable elements highlighting the relationship between genome position and gene evolution in this pathogen species. Conclusion. Our comparative genomics analyses of host-specific M. oryzae isolates revealed gain and loss of genes as a major evolutionary mechanism driving specialization to Oryza and Setaria. Transposable elements appear to facilitate gene evolution possibly by enhancing chromosomal rearrangements and other forms of genetic variation.
|
|
Scooped by
Kamoun Lab @ TSL
April 5, 2016 3:49 AM
|
Sequencing unsampled fungal diversity
|
|
Scooped by
Kamoun Lab @ TSL
October 21, 2015 9:28 AM
|
Background. Downy mildews are the most speciose group of oomycetes and affect crops of great economic importance. So far, there is only a single deeply-sequenced downy mildew genome available, from Hyaloperonospora arabidopsidis. Further genomic resources for downy mildews are required to study their evolution, including pathogenicity effector proteins, such as RxLR effectors. Plasmopara halstedii is a devastating pathogen of sunflower and a potential pathosystem model to study downy mildews, as several Avr-genes and R-genes have been predicted and unlike Arabidopsis downy mildew, large quantities of almost contamination-free material can be obtained easily.
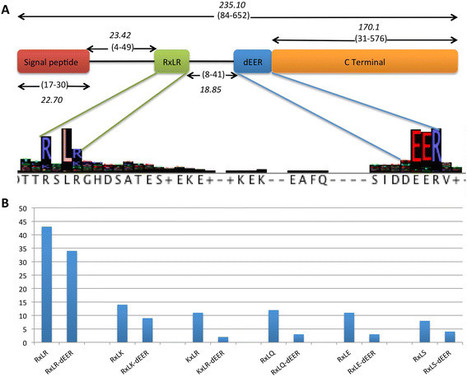
Results. Here a high-quality draft genome of Plasmopara halstedii is reported and analysed with respect to various aspects, including genome organisation, secondary metabolism, effector proteins and comparative genomics with other sequenced oomycetes. Interestingly, the present analyses revealed further variation of the RxLR motif, suggesting an important role of the conservation of the dEER-motif. Orthology analyses revealed the conservation of 28 RxLR-like core effectors among Phytophthora species. Only six putative RxLR-like effectors were shared by the two sequenced downy mildews, highlighting the fast and largely independent evolution of two of the three major downy mildew lineages. This is seemingly supported by phylogenomic results, in which downy mildews did not appear to be monophyletic.
Conclusions. The genome resource will be useful for developing markers for monitoring the pathogen population and might provide the basis for new approaches to fight Phytophthora and downy mildew pathogens by targeting core pathogenicity effectors.
|
|
Rescooped by
Kamoun Lab @ TSL
from Plants and Microbes
September 4, 2015 6:04 AM
|
- Development of resistant crops is the most effective way to control plant diseases to safeguard food and feed production. Disease resistance is commonly based on resistance genes, which generally mediate the recognition of small proteins secreted by invading pathogens. These proteins secreted by pathogens are called ‘avirulence’ proteins. Their identification is important for being able to assess the usefulness and durability of resistance genes in agricultural settings.
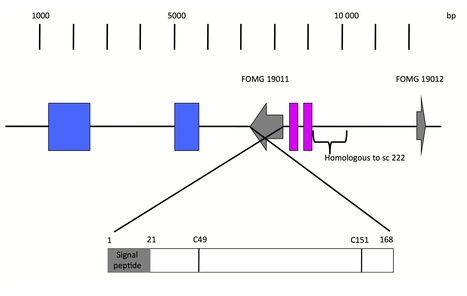
- We have used genome sequencing of a set of strains of the melon wilt fungus Fusarium oxysporum f. sp. melonis (Fom), bioinformatics-based genome comparison and genetic transformation of the fungus to identify AVRFOM2, the gene that encodes the avirulence protein recognized by the melon Fom-2 gene.
- Both an unbiased and a candidate gene approach identified a single candidate for the AVRFOM2 gene. Genetic complementation of AVRFOM2 in three different race 2 isolates resulted in resistance of Fom-2-harbouring melon cultivars. AvrFom2 is a small, secreted protein with two cysteine residues and weak similarity to secreted proteins of other fungi.
- The identification of AVRFOM2 will not only be helpful to select melon cultivars to avoid melon Fusarium wilt, but also to monitor how quickly a Fom population can adapt to deployment of Fom-2-containing cultivars in the field.
|
|
|
Scooped by
Kamoun Lab @ TSL
February 13, 2017 4:08 AM
|
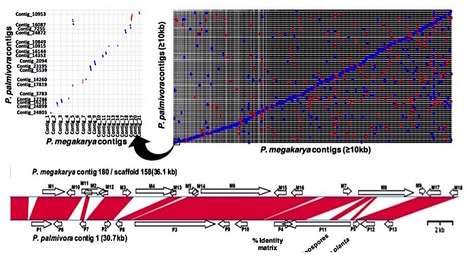
Phytophthora megakarya (Pmeg) and P. palmivora (Ppal) are closely related species causing cacao black pod rot. While Ppal is a cosmopolitan pathogen, cacao is the only known host of economic importance for Pmeg. Pmeg is more virulent on cacao than Ppal. We sequenced and compared the Pmeg and Ppal genomes and identified virulence-related putative gene models (PGeneM) that may be responsible for their differences in host specificities and virulence. Pmeg and Ppal have estimated genome sizes of 126.88 and 151.23 Mb and PGeneM numbers of 42,036 and 44,327, respectively. The evolutionary histories of Pmeg and Ppal appear quite different. Post-speciation, Ppal underwent whole-genome duplication whereas Pmeg has undergone selective increases in PGeneM numbers, likely through accelerated transposable element driven duplications. Many PGeneMs in both species failed to match transcripts and may represent pseudogenes or cryptic genetic reservoirs. Pmeg appears to have amplified specific gene families, some of which are virulence-related. Analysis of mycelium, zoospore and in planta transcriptome expression profiles using neural network self-organizing map analysis generated 24 multi-variate and non-linear self-organizing map classes. Many members of the RxLR, NPP, and pectinase genes families were specifically induced in planta. Pmeg displays a diverse virulence-related gene complement similar in size to and potentially of greater diversity than Ppal but it remains likely that the specific functions of the genes determine each species’ unique characteristics as pathogens.
|
|
Scooped by
Kamoun Lab @ TSL
December 26, 2016 6:06 AM
|
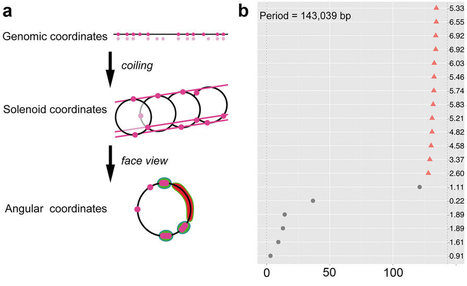
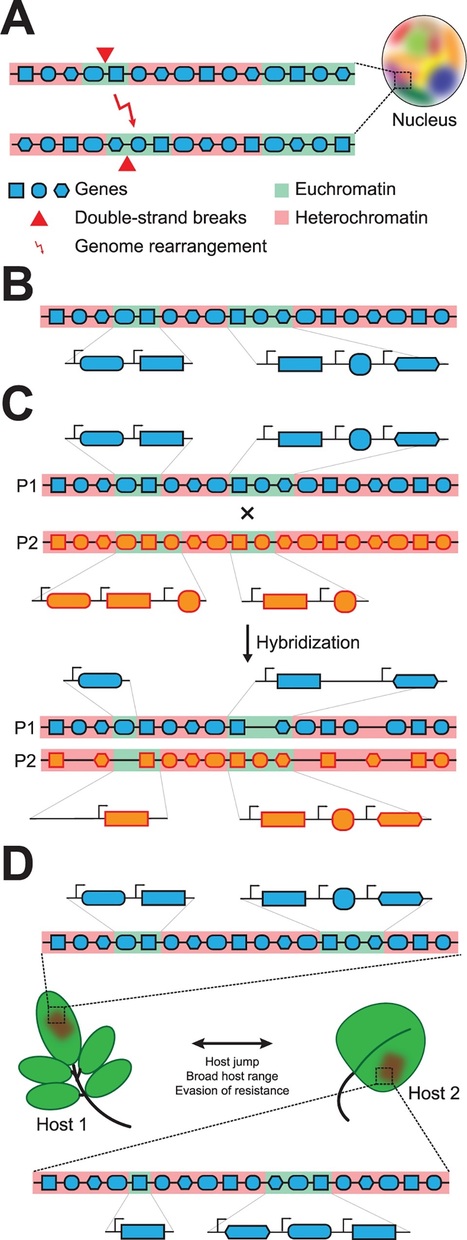
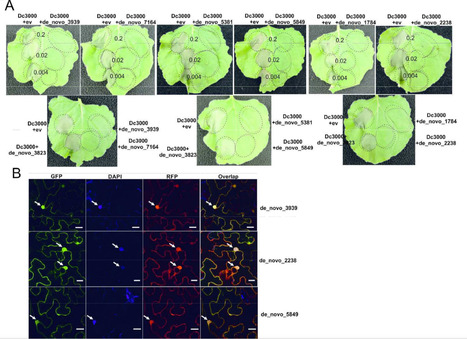
An organism’s adaptation to changing environments is fueled by its genetic variability, which is established by mechanisms ranging from single-nucleotide polymorphisms to large-scale structural variations, all of which affect chromosomal shape, organization, and gene content [1]. These processes are particularly relevant for pathogens that must respond to continual selection pressure arising from host immune systems that evolved to detect the presence or activity of potential microbial pathogens through a variety of invasion patterns [2]. In their adaptive response, pathogens evolve strategies, often involving secreted effector molecules, to overcome host immunity and support host colonization [3]. Thus, it can be anticipated that this coevolutionary arms race leads to highly specific interactions between adapted pathogens and their specific hosts. Paradoxically, particular pathogens successfully colonize a broad range of hosts, yet how such pathogens cope in arms races with such a diversity of hosts remains unknown. A structured genome drives adaptive evolution. It has been proposed that filamentous fungal and oomycete plant pathogens often evolved structured genomes with two distinct types of genomic regions: (1) gene-rich regions containing slowly evolving genes that mediate general physiology and (2) gene-poor regions that are dynamic and enriched for repetitive DNA such as transposable elements (TEs) and virulence-related genes that often display signs of accelerated evolution [4,5]. Extensive structural variation often occurs in these fast-evolving regions, leading to translocation, duplication, or loss of genetic material [1,4,5]. The highly dynamic regions can either be embedded within the core chromosomes or be located on separate chromosomes that often display presence/absence variations within a population, known as conditionally dispensable or accessory chromosomes [4,6]. The common occurrence of these bipartite genomes led to the “two-speed” model for pathogen genome evolution [4], suggesting that specific regions form sites of rapid genomic diversification to facilitate coevolution during host interactions [1,4,5]. Transposable elements shape “two-speed” genomes. It is generally observed that the dynamic regions of two-speed genomes are enriched in TEs, yet it remains undemonstrated how they mechanistically contribute to genome variability [1,4,5]. Recently, the contribution of TEs towards the evolution of the two-speed genome in the vascular wilt pathogen Verticillium dahliae was reported [7]. Extensive genome rearrangements are generated by double-strand repair pathways that erroneously utilize stretches of homologous sequence at an unlinked locus, the majority of which occur around TEs simply as a consequence of their abundance and sequence similarity (Fig 1A) [7]. Genomic rearrangements often occur at dynamic regions that are enriched for recent segmental duplications, generating genetic material subject to evolutionary diversification, e.g., by reciprocal gene loss (Fig 1A) [7,8]. Furthermore, these dynamic regions are enriched in in planta induced effectors [8] and evolutionary young and “active” TEs (Fig 1B). These TEs likely contribute to accelerated genomic diversification of dynamic effector regions [7].
|
|
Scooped by
Kamoun Lab @ TSL
November 23, 2016 6:38 AM
|
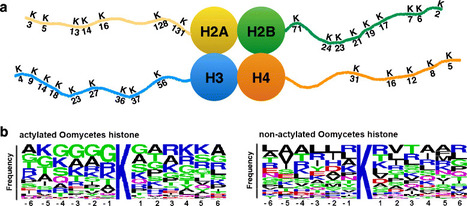
Background - Oomycetes are a group of fungus-like eukaryotes with diverse microorganisms living in marine, freshwater and terrestrial environments. Many of them are important pathogens of plants and animals, causing severe economic losses. Based on previous study, gene expression in eukaryotic cells is regulated by epigenetic mechanisms such as DNA methylation and histone modification. However, little is known about epigenetic mechanisms of oomycetes. Results - In this study, we investigated the candidate genes in regulating histone acetylation in oomycetes genomes through bioinformatics approaches and identified a group of diverse histone acetyltransferases (HATs) and histone deacetylases (HDACs), along with three putative novel HATs. Phylogenetic analyses suggested that most of these oomycetes HATs and HDACs derived from distinct evolutionary ancestors. Phylogenetic based analysis revealed the complex and distinct patterns of duplications and losses of HATs and HDACs in oomycetes. Moreover, gene expression analysis unveiled the specific expression patterns of the 33 HATs and 11 HDACs of Phytophthora infestans during the stages of development, infection and stress response. Conclusions - In this study, we reveal the structure, diversity and the phylogeny of HATs and HDACs of oomycetes. By analyzing the expression data, we provide an overview of the specific biological stages of these genes involved. Our datasets provide useful inputs to help explore the epigenetic mechanisms and the relationship between genomes and phenotypes of oomycetes.
|
|
Scooped by
Kamoun Lab @ TSL
November 2, 2016 8:08 AM
|
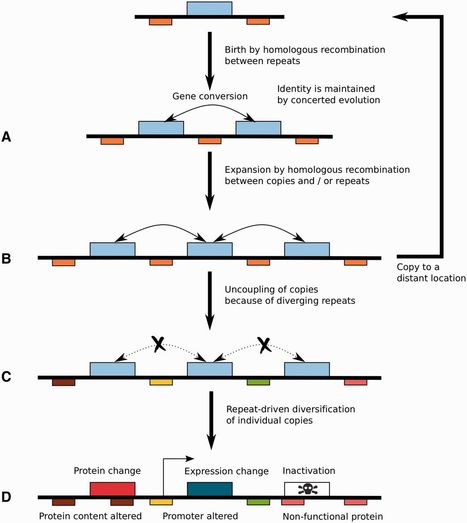
Smut fungi are plant pathogens mostly parasitizing wild species of grasses as well as domesticated cereal crops. Genome analysis of several smut fungi including Ustilago maydis revealed a singular clustered organization of genes encoding secreted effectors. In U. maydis, many of these clusters have a role in virulence. Reconstructing the evolutionary history of clusters of effector genes is difficult because of their intrinsically fast evolution, which erodes the phylogenetic signal and homology relationships. Here, we describe the use of comparative evolutionary analyses of quality draft assemblies of genomes to study the mechanisms of this evolution. We report the genome sequence of a South African isolate of Sporisorium scitamineum, a smut fungus parasitizing sugar cane with a phylogenetic position intermediate to the two previously sequenced species U. maydisand Sporisorium reilianum. We show that the genome of S. scitamineumcontains more and larger gene clusters encoding secreted effectors than any previously described species in this group. We trace back the origin of the clusters and find that their evolution is mainly driven by tandem gene duplication. In addition, transposable elements play a major role in the evolution of the clustered genes. Transposable elements are significantly associated with clusters of genes encoding fast evolving secreted effectors. This suggests that such clusters represent a case of genome compartmentalization that restrains the activity of transposable elements on genes under diversifying selection for which this activity is potentially beneficial, while protecting the rest of the genome from its deleterious effect.
|
|
Scooped by
Kamoun Lab @ TSL
September 9, 2016 6:09 AM
|
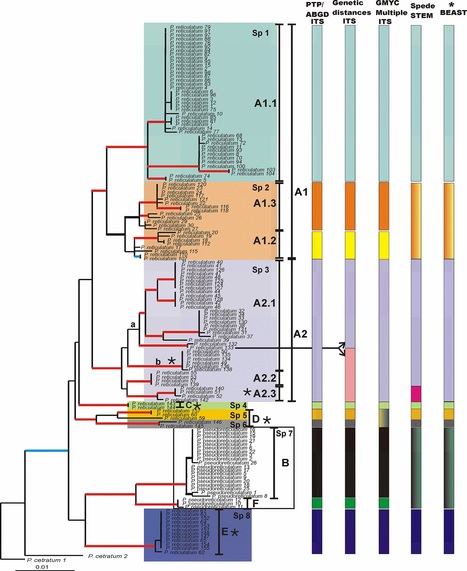
Asexual species with vegetative propagation of both symbiont partners (soredia) in lichens may harbor lower species diversity because they may indeed represent evolutionary dead ends or clones. In this study we aim to critically examine species boundaries in the sorediate lichen forming fungi Parmotrema reticulatum–Parmotrema pseudoreticulatum complex applying coalescent-based approaches and other recently developed DNA-based methods. To this end, we gathered 180 samples from Africa, Asia, Australasia, Europe, North and South America and generated sequences of internal transcribed spacer of nuclear ribosomal DNA (ITS) and DNA replication licensing factor MCM7 (MCM7). The dataset was analysed using different approaches such as traditional phylogeny–maximum likelihood and Bayesian–genetic distances, automatic barcode gap discovery and coalescent-based methods–PTP, GMYC, spedeSTEM and *Beast–in order to test congruence among results. Additionally, the divergence times were also estimated to elucidate diversification events. Delimitations inferred from the different analyses are comparable with only minor differences, and following a conservative approach we propose that the sampled specimens of the P. reticulatum–P. pseudoreticulatum complex belong to at least eight distinct species-level lineages. Seven are currently classified under P. reticulatum and one as P. pseudoreticulatum. In this work we discuss one of only few examples of cryptic species that have so far been found in sorediate reproducing lichen forming fungi. Additionally our estimates suggest a recent origin of the species complex–during the Miocene. Consequently, the wide distribution of several of the cryptic species has to be explained by intercontinental long-distance dispersal events.
|
|
Scooped by
Kamoun Lab @ TSL
September 5, 2016 6:56 AM
|
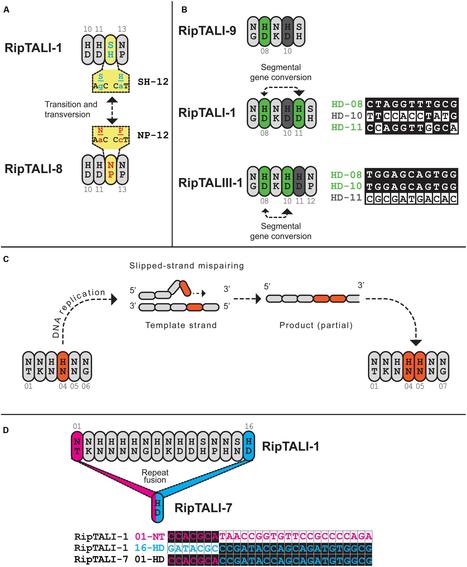
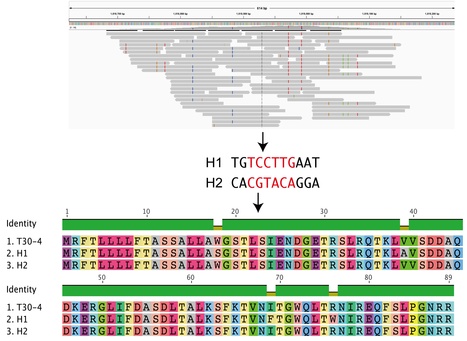
Ralstonia solanacearum, a species complex of bacterial plant pathogens divided into four monophyletic phylotypes, causes plant diseases in tropical climates around the world. Some strains exhibit a broad host range on solanaceous hosts, while others are highly host-specific as for example some banana-pathogenic strains. Previous studies showed that transcription activator-like (TAL) effectors from Ralstonia, termed RipTALs, are capable of activating reporter genes in planta, if these are preceded by a matching effector binding element (EBE). RipTALs target DNA via their central repeat domain (CRD), where one repeat pairs with one DNA-base of the given EBE. The repeat variable diresidue dictates base repeat specificity in a predictable fashion, known as the TALE code. In this work, we analyze RipTALs across all phylotypes of the Ralstonia solanacearum species complex. We find that RipTALs are prevalent in phylotypes I and IV but absent from most phylotype III and II strains (10/12, 8/14, 1/24, and 1/5 strains contained a RipTAL, respectively). RipTALs originating from strains of the same phylotype show high levels of sequence similarity (>98%) in the N-terminal and C-terminal regions, while RipTALs isolated from different phylotypes show 47–91% sequence similarity in those regions, giving rise to four RipTAL classes. We show that, despite sequence divergence, the base preference for guanine, mediated by the N-terminal region, is conserved across RipTALs of all classes. Using the number and order of repeats found in the CRD, we functionally sub-classify RipTALs, introduce a new simple nomenclature, and predict matching EBEs for all seven distinct RipTALs identified. We experimentally study RipTAL EBEs and uncover that some RipTALs are able to target the EBEs of other RipTALs, referred to as cross-reactivity. In particular, RipTALs from strains with a broad host range on solanaceous hosts cross-react on each other’s EBEs. Investigation of sequence divergence between RipTAL repeats allows for a reconstruction of repeat array biogenesis, for example through slipped strand mispairing or gene conversion. Using these studies we show how RipTALs of broad host range strains evolved convergently toward a shared target sequence. Finally, we discuss the differences between TALE-likes of plant pathogens in the context of disease ecology.
|
|
Scooped by
Kamoun Lab @ TSL
September 1, 2016 9:32 AM
|
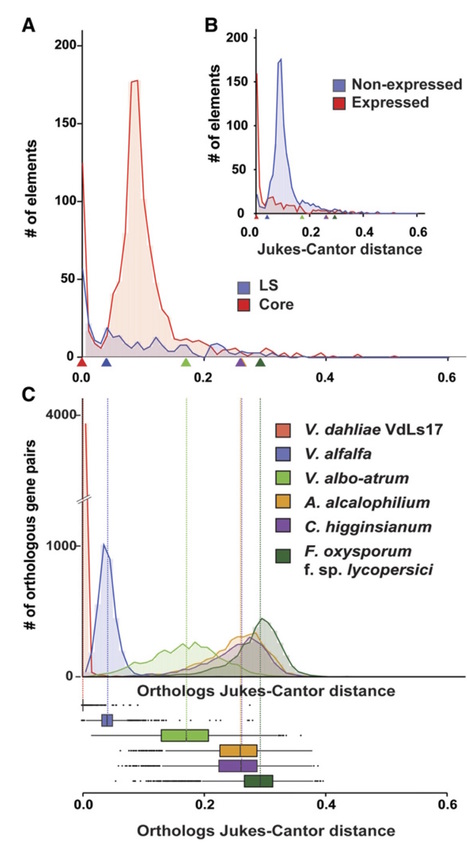
Genomic plasticity enables adaptation to changing environments, which is especially relevant for pathogens that engage in 'arms races' with their hosts. In many pathogens, genes mediating virulence cluster in highly variable, transposon-rich, physically distinct genomic compartments. However, understanding of the evolution of these compartments, and the role of transposons therein, remains limited. Here, we show that transposons are the major driving force for adaptive genome evolution in the fungal plant pathogen Verticillium dahliae. We show that highly variable lineage-specific (LS) regions evolved by genomic rearrangements that are mediated by erroneous double-strand repair, often utilizing transposons. We furthermore show that recent genetic duplications are enhanced in LS regions, against an older episode of duplication events. Finally, LS regions are enriched in active transposons, which contribute to local genome plasticity. Thus, we provide evidence for genome shaping by transposons, both in an active and passive manner, which impacts the evolution of pathogen virulence.
|
|
Scooped by
Kamoun Lab @ TSL
June 28, 2016 8:27 AM
|
Three members of the Puccini genus, P. triticina (Pt), P. striiformis f.sp. tritici (Pst), and P. graminis f.sp. tritici (Pgt), cause the most common and often most significant foliar diseases of wheat. While similar in biology and life cycle, each species is uniquely adapted and specialized. The genomes of Pt and Pst were sequenced and compared to that of Pgt to identify common and distinguishing gene content, to determine gene variation among wheat rust pathogens, other rust fungi and basidiomycetes, and to identify genes of significance for infection. Pt had the largest genome of the three, estimated at 135 Mb with expansion due to mobile elements and repeats encompassing 50.9% of contig bases; by comparison repeats occupy 31.5% for Pst and 36.5% for Pgt. We find all three genomes are highly heterozygous, with Pst (5.97 SNPs/kb) nearly twice the level detected in Pt (2.57 SNPs/kb) and that previously reported for Pgt. Of 1,358 predicted effectors in Pt, 784 were found expressed across diverse life cycle stages including the sexual stage. Comparison to related fungi highlighted the expansion of gene families involved in transcriptional regulation and nucleotide binding, protein modification, and carbohydrate enzyme degradation. Two allelic homeodomain, HD1 and HD2, pairs and three pheromone receptor (STE3) mating-type genes were identified in each dikaryotic Pucciniaspecies. The HD proteins were active in a heterologous Ustilago maydis mating assay and host induced gene silencing of the HD and STE3 alleles reduced wheat host infection.
|
|
Scooped by
Kamoun Lab @ TSL
May 24, 2016 9:22 AM
|
Background. Understanding how plants and pathogens modulate gene expression during the host-pathogen interaction is key to uncovering the molecular mechanisms that regulate disease progression. Recent advances in sequencing technologies have provided new opportunities to decode the complexity of such interactions. In this study, we used an RNA-based sequencing approach (RNA-seq) to assess the global expression profiles of the wheat yellow rust pathogen Puccinia striiformis f. sp. tritici (PST) and its host during infection. Results. We performed a detailed RNA-seq time-course for a susceptible and a resistant wheat host infected with PST. This study (i) defined the global gene expression profiles for PST and its wheat host, (ii) substantially improved the gene models for PST, (iii) evaluated the utility of several programmes for quantification of global gene expression for PST and wheat, and (iv) identified clusters of differentially expressed genes in the host and pathogen. By focusing on components of the defence response in susceptible and resistant hosts, we were able to visualise the effect of PST infection on the expression of various defence components and host immune receptors. Conclusions. Our data showed sequential, temporally coordinated activation and suppression of expression of a suite of immune-response regulators that varied between compatible and incompatible interactions. These findings provide the framework for a better understanding of how PST causes disease and support the idea that PST can suppress the expression of defence components in wheat to successfully colonize a susceptible host.
|
|
Scooped by
Kamoun Lab @ TSL
May 5, 2016 6:22 AM
|
Background - The protist Plasmodiophora brassicae is a soil-borne pathogen of cruciferous species and the causal agent of clubroot disease of Brassicas including agriculturally important crops such as canola/rapeseed (Brassica napus). P. brassicae has remained an enigmatic plant pathogen and is a rare example of an obligate biotroph that resides entirely inside the host plant cell. The pathogen is the cause of severe yield losses and can render infested fields unsuitable for Brassica crop growth due to the persistence of resting spores in the soil for up to 20 years.
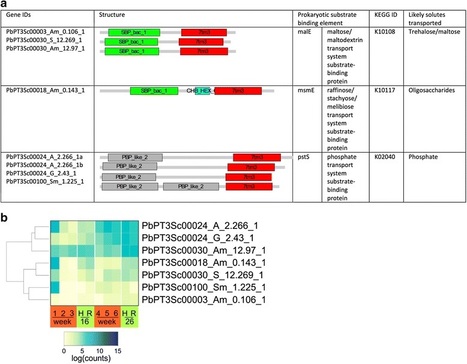
Results - To provide insight into the biology of the pathogen and its interaction with its primary host B. napus, we produced a draft genome of P. brassicae pathotypes 3 and 6 (Pb3 and Pb6) that differ in their host range. Pb3 is highly virulent on B. napus (but also infects other Brassica species) while Pb6 infects only vegetable Brassica crops. Both the Pb3 and Pb6 genomes are highly compact, each with a total size of 24.2 Mb, and contain less than 2 % repetitive DNA. Clustering of genome-wide single nucleotide polymorphisms (SNP) of Pb3, Pb6 and three additional re-sequenced pathotypes (Pb2, Pb5 and Pb8) shows a high degree of correlation of cluster grouping with host range. The Pb3 genome features significant reduction of intergenic space with multiple examples of overlapping untranslated regions (UTRs). Dependency on the host for essential nutrients is evident from the loss of genes for the biosynthesis of thiamine and some amino acids and the presence of a wide range of transport proteins, including some unique to P. brassicae. The annotated genes of Pb3 include those with a potential role in the regulation of the plant growth hormones cytokinin and auxin. The expression profile of Pb3 genes, including putative effectors, during infection and their potential role in manipulation of host defence is discussed.
Conclusion - The P. brassicae genome sequence reveals a compact genome, a dependency of the pathogen on its host for some essential nutrients and a potential role in the regulation of host plant cytokinin and auxin. Genome annotation supported by RNA sequencing reveals significant reduction in intergenic space which, in addition to low repeat content, has likely contributed to the P. brassicae compact genome.
|
|
Scooped by
Kamoun Lab @ TSL
March 31, 2016 11:06 AM
|
Asian soybean rust (ASR) caused by the obligate biotrophic fungus Phakopsora pachyrhizi can cause losses greater than 80%. Despite its economic importance, there is no soybean cultivar with durable ASR resistance (Goellner et al., 2010). In addition, the P. pachyrhizi genome is not yet available. However, the availability of other rust genomes as well as the development of sample enrichment strategies and bioinformatics tools has improved our knowledge of the ASR secretome and its potential effectors. The authors used a combination of laser capture microdissection (LCM), RNAseq and a bioinformatics pipeline to identify a total of 36,350 P. pachyrhizi contigs expressed in planta and a predicted secretome of 851 proteins.
|
|
Scooped by
Kamoun Lab @ TSL
September 8, 2015 7:26 AM
|
The application of DNA sequencing technology to the study of ancient DNA has enabled the reconstruction of past epidemics from genomes of historically important plant-associated microbes. Recently, the genome sequences of the potato late blight pathogen Phytophthora infestans were analyzed from 19th century herbarium specimens. These herbarium samples originated from infected potatoes collected during and after the Irish potato famine. Herbaria have therefore great potential to help elucidate past epidemics of crops, date the emergence of pathogens, and inform about past pathogen population dynamics. DNA preservation in herbarium samples was unexpectedly good, raising the possibility of a whole new research area in plant and microbial genomics. However, the recovered DNA can be extremely fragmented resulting in specific challenges in reconstructing genome sequences. Here we review some of the challenges in computational analyses of ancient DNA from herbarium samples. We also applied the recently developed linkage method to haplotype reconstruction of diploid or polyploid genomes from fragmented ancient DNA.
|