Your new post is loading...
Your new post is loading...

|
Scooped by
Kamoun Lab @ TSL
October 11, 2011 12:36 PM
|
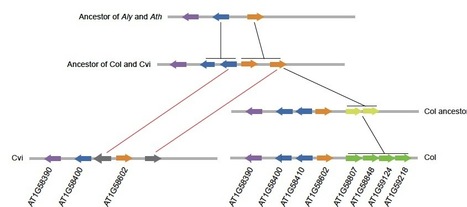
Plants, like animals, use several lines of defense against pathogen attack. Prominent among genes that confer disease resistance are those encoding nucleotide binding site-leucine rich repeat (NB-LRR) proteins. Likely due to selection pressures caused by pathogens, NB-LRR genes are the most variable gene family in plants, but there appear to be species-specific limits to the number of NB-LRR genes in a genome. Allelic diversity within an individual is also increased by obligatory outcrossing, which leads to genome-wide heterozygosity. In the present study, we therefore compared the NB-LRR gene complement of the selfer Arabidopsis thaliana with its outcrossing close relative A. lyrata. We then complemented and contrasted the interspecific patterns with studies of NB-LRR diversity within A. thaliana. Three important insights are (i) that both species have similar numbers of NB-LRR genes; (ii) that loci with single NB-LRR genes are less variable than tandem arrays; and (iii) that presence-absence polymorphisms within A. thaliana are not strongly correlated with the presence or absence of orthologs in A. lyrata. Although A. thaliana individuals are mostly homozygous and thus potentially less likely to suffer from aberrant interaction of NB-LRR proteins with newly introduced alleles, the number of NB-LRR genes is similar to that in A. lyrata. In intra-and interspecific comparisons, NB-LRR genes are also more variable than Receptor-Like Protein (RLPs) genes. Finally, in contrast to Drosophila, there is a clearly positive relationship between interspecific divergence and intraspecific polymorphisms.
|
|
Scooped by
Kamoun Lab @ TSL
September 21, 2011 5:11 AM
|
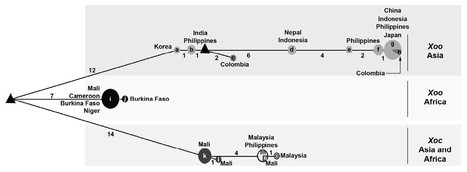
Multilocus sequence analysis (MLSA) and type III effector (T3E) repertoire mining were performed to gain new insights into the genetic relatedness of Xanthomonas oryzae pv. oryzae (Xoo) and Xanthomonas oryzae pv. oryzicola (Xoc), two major bacterial pathogens of rice. Based on a collection of 45 African and Asian strains, we first sequenced and analysed three housekeeping genes by MLSA, Bayesian clustering and a median-joining network approach. Second, we investigated the distribution of 32 T3E genes, which are known to be major virulence factors of plant pathogenic bacteria, in all selected strains, by polymerase chain reaction and dot-blot hybridization methods. The diversity observed within housekeeping genes, as well as within T3E repertoires, clearly showed that both pathogens belong to closely related, but distinct, phylogenetic groups. Interestingly, these evolutionary groups are differentiated according to the geographical origin of the strains, suggesting that populations of Xoo and Xoc might be endemic in Africa and Asia, and thus have evolved separately. We further revealed that T3E gene repertoires of both pathogens comprise core and variable gene suites that probably have distinct roles in pathogenicity and different evolutionary histories. In this study, we carried out a functional analysis of xopO, a differential T3E gene between Xoo and Xoc, to determine the involvement of this gene in tissue specificity. Altogether, our data contribute to a better understanding of the evolutionary history of Xoo and Xoc in Africa and Asia, and provide clues for functional studies aiming to understand the virulence, host and tissue specificity of both rice pathogens.
|
|
Scooped by
Kamoun Lab @ TSL
September 8, 2011 11:01 AM
|
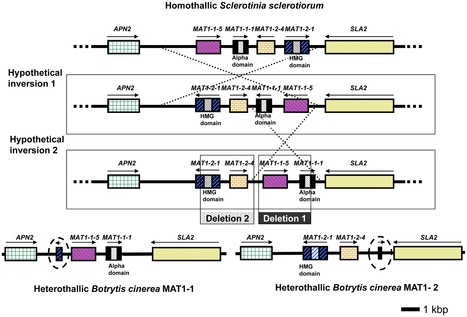
Sclerotinia sclerotiorum and Botrytis cinerea are closely related necrotrophic plant pathogenic fungi notable for their wide host ranges and environmental persistence. These attributes have made these species models for understanding the complexity of necrotrophic, broad host-range pathogenicity. Despite their similarities, the two species differ in mating behaviour and the ability to produce asexual spores. We have sequenced the genomes of one strain of S. sclerotiorum and two strains of B. cinerea. The comparative analysis of these genomes relative to one another and to other sequenced fungal genomes is provided here. Their 38–39 Mb genomes include 11,860–14,270 predicted genes, which share 83% amino acid identity on average between the two species. We have mapped the S. sclerotiorum assembly to 16 chromosomes and found large-scale co-linearity with the B. cinerea genomes. Seven percent of the S. sclerotiorum genome comprises transposable elements compared to <1% of B. cinerea. The arsenal of genes associated with necrotrophic processes is similar between the species, including genes involved in plant cell wall degradation and oxalic acid production. Analysis of secondary metabolism gene clusters revealed an expansion in number and diversity of B. cinerea–specific secondary metabolites relative to S. sclerotiorum. The potential diversity in secondary metabolism might be involved in adaptation to specific ecological niches. Comparative genome analysis revealed the basis of differing sexual mating compatibility systems between S. sclerotiorum and B. cinerea. The organization of the mating-type loci differs, and their structures provide evidence for the evolution of heterothallism from homothallism. These data shed light on the evolutionary and mechanistic bases of the genetically complex traits of necrotrophic pathogenicity and sexual mating. This resource should facilitate the functional studies designed to better understand what makes these fungi such successful and persistent pathogens of agronomic crops.
|
|
Scooped by
Kamoun Lab @ TSL
September 2, 2011 8:34 PM
|
Horizontal gene transfer (HGT) can radically alter the genomes of microorganisms, providing the capacity to adapt to new lifestyles, environments, and hosts. However, the extent of HGT between eukaryotes is unclear. Using whole-genome, gene-by-gene phylogenetic analysis we demonstrate an extensive pattern of cross-kingdom HGT between fungi and oomycetes. Comparative genomics, including the de novo genome sequence of Hyphochytrium catenoides, a free-living sister of the oomycetes, shows that these transfers largely converge within the radiation of oomycetes that colonize plant tissues. The repertoire of HGTs includes a large number of putatively secreted proteins; for example, 7.6% of the secreted proteome of the sudden oak death parasite Phytophthora ramorum has been acquired from fungi by HGT. Transfers include gene products with the capacity to break down plant cell walls and acquire sugars, nucleic acids, nitrogen, and phosphate sources from the environment. Predicted HGTs also include proteins implicated in resisting plant defense mechanisms and effector proteins for attacking plant cells. These data are consistent with the hypothesis that some oomycetes became successful plant parasites by multiple acquisitions of genes from fungi.
|
|
Scooped by
Kamoun Lab @ TSL
August 23, 2011 7:16 PM
|
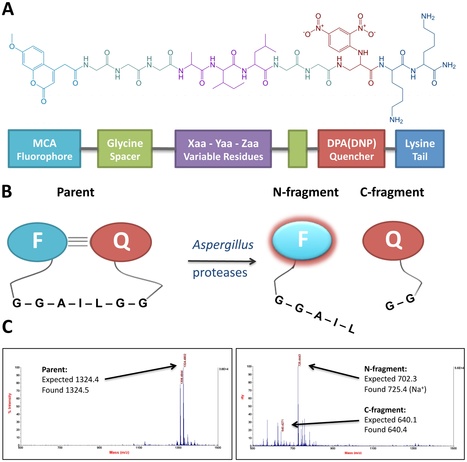
Background - The filamentous fungus Aspergillus fumigatus (AF) can cause devastating infections in immunocompromised individuals. Early diagnosis improves patient outcomes but remains challenging because of the limitations of current methods. To augment the clinician's toolkit for rapid diagnosis of AF infections, we are investigating AF secreted proteases as novel diagnostic targets. The AF genome encodes up to 100 secreted proteases, but fewer than 15 of these enzymes have been characterized thus far. Given the large number of proteases in the genome, studies focused on individual enzymes may overlook potential diagnostic biomarkers. Methodology and Principal Findings - As an alternative, we employed a combinatorial library of internally quenched fluorogenic probes (IQFPs) to profile the global proteolytic secretome of an AF clinical isolate in vitro. Comparative protease activity profiling revealed 212 substrate sequences that were cleaved by AF secreted proteases but not by normal human serum. A central finding was that isoleucine, leucine, phenylalanine, and tyrosine predominated at each of the three variable positions of the library (44.1%, 59.1%, and 57.0%, respectively) among substrate sequences cleaved by AF secreted proteases. In contrast, fewer than 10% of the residues at each position of cleaved sequences were cationic or anionic. Consensus substrate motifs were cleaved by thermostable serine proteases that retained activity up to 50°C. Precise proteolytic cleavage sites were reliably determined by a simple, rapid mass spectrometry-based method, revealing predominantly non-prime side specificity. A comparison of the secreted protease activities of three AF clinical isolates revealed consistent protease substrate specificity fingerprints. However, secreted proteases of A. flavus, A. nidulans, and A. terreus strains exhibited striking differences in their proteolytic signatures. Conclusions - This report provides proof-of-principle for the use of protease substrate specificity profiling to define the proteolytic secretome of Aspergillus fumigatus. Expansion of this technique to protease secretion during infection could lead to development of novel approaches to fungal diagnosis.
|
|
Scooped by
Kamoun Lab @ TSL
August 5, 2011 6:44 AM
|
Brown rot decay removes cellulose and hemicellulose from wood—residual lignin contributing up to 30% of forest soil carbon—and is derived from an ancestral white rot saprotrophy in which both lignin and cellulose are decomposed. Comparative and functional genomics of the “dry rot” fungus Serpula lacrymans, derived from forest ancestors, demonstrated that the evolution of both ectomycorrhizal biotrophy and brown rot saprotrophy were accompanied by reductions and losses in specific protein families, suggesting adaptation to an intercellular interaction with plant tissue. Transcriptome and proteome analysis also identified differences in wood decomposition in S. lacrymans relative to the brown rot Postia placenta. Furthermore, fungal nutritional mode diversification suggests that the boreal forest biome originated via genetic coevolution of above- and below-ground biota.
|
|
Scooped by
Kamoun Lab @ TSL
August 1, 2011 4:56 PM
|
The vascular wilt fungi Verticillium dahliae and V. albo-atrum infect over 200 plant species, causing billions of dollars in annual crop losses. The characteristic wilt symptoms are a result of colonization and proliferation of the pathogens in the xylem vessels, which undergo fluctuations in osmolarity. To gain insights into the mechanisms that confer the organisms' pathogenicity and enable them to proliferate in the unique ecological niche of the plant vascular system, we sequenced the genomes of V. dahliae and V. albo-atrum and compared them to each other, and to the genome of Fusarium oxysporum, another fungal wilt pathogen. Our analyses identified a set of proteins that are shared among all three wilt pathogens, and present in few other fungal species. One of these is a homolog of a bacterial glucosyltransferase that synthesizes virulence-related osmoregulated periplasmic glucans in bacteria. Pathogenicity tests of the corresponding V. dahliae glucosyltransferase gene deletion mutants indicate that the gene is required for full virulence in the Australian tobacco species Nicotiana benthamiana. Compared to other fungi, the two sequenced Verticillium genomes encode more pectin-degrading enzymes and other carbohydrate-active enzymes, suggesting an extraordinary capacity to degrade plant pectin barricades. The high level of synteny between the two Verticillium assemblies highlighted four flexible genomic islands in V. dahliae that are enriched for transposable elements, and contain duplicated genes and genes that are important in signaling/transcriptional regulation and iron/lipid metabolism. Coupled with an enhanced capacity to degrade plant materials, these genomic islands may contribute to the expanded genetic diversity and virulence of V. dahliae, the primary causal agent of Verticillium wilts. Significantly, our study reveals insights into the genetic mechanisms of niche adaptation of fungal wilt pathogens, advances our understanding of the evolution and development of their pathogenesis, and sheds light on potential avenues for the development of novel disease management strategies to combat destructive wilt diseases.
|
|
Scooped by
Kamoun Lab @ TSL
July 25, 2011 6:24 AM
|
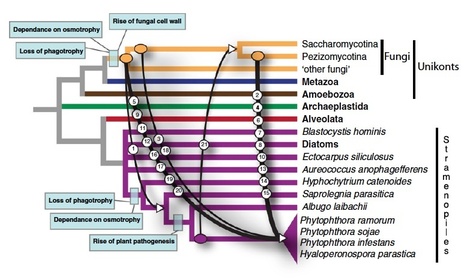
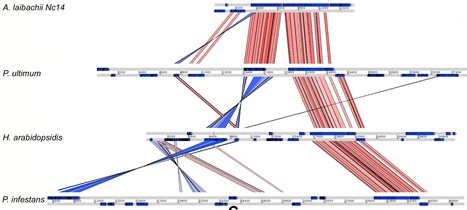
Biotrophic eukaryotic plant pathogens require a living host for their growth and form an intimate haustorial interface with parasitized cells. Evolution to biotrophy occurred independently in fungal rusts and powdery mildews, and in oomycete white rusts and downy mildews. Biotroph evolution and molecular mechanisms of biotrophy are poorly understood. It has been proposed, but not shown, that obligate biotrophy results from (i) reduced selection for maintenance of biosynthetic pathways and (ii) gain of mechanisms to evade host recognition or suppress host defence. Here we use Illumina sequencing to define the genome, transcriptome, and gene models for the obligate biotroph oomycete and Arabidopsis parasite, Albugo laibachii. A. laibachii is a member of the Chromalveolata, which incorporates Heterokonts (containing the oomycetes), Apicomplexa (which includes human parasites like Plasmodium falciparum and Toxoplasma gondii), and four other taxa. From comparisons with other oomycete plant pathogens and other chromalveolates, we reveal independent loss of molybdenum-cofactor-requiring enzymes in downy mildews, white rusts, and the malaria parasite P. falciparum. Biotrophy also requires “effectors” to suppress host defence; we reveal RXLR and Crinkler effectors shared with other oomycetes, and also discover and verify a novel class of effectors, the “CHXCs”, by showing effector delivery and effector functionality. Our findings suggest that evolution to progressively more intimate association between host and parasite results in reduced selection for retention of certain biosynthetic pathways, and particularly reduced selection for retention of molybdopterin-requiring biosynthetic pathways. These mechanisms are not only relevant to plant pathogenic oomycetes but also to human pathogens within the Chromalveolata.
|
|
Scooped by
Kamoun Lab @ TSL
July 25, 2011 6:17 AM
|
Background - Gene loss, inversions, translocations, and other chromosomal rearrangements vary among species, resulting in different rates of structural genome evolution. Major chromosomal rearrangements are rare in most eukaryotes, giving large regions with the same genes in the same order and orientation across species. These regions of macrosynteny have been very useful for locating homologous genes in different species and to guide the assembly of genome sequences. Previous analyses in the fungi have indicated that macrosynteny is rare; instead, comparisons across species show no synteny or only microsyntenic regions encompassing usually five or fewer genes. To test the hypothesis that chromosomal evolution is different in the fungi compared to other eukaryotes, synteny was compared between species of the major fungal taxa.
Results - These analyses identified a novel form of evolution in which genes are conserved within homologous chromosomes, but with randomized orders and orientations. This mode of evolution is designated mesosynteny, to differentiate it from micro- and macrosynteny seen in other organisms. Mesosynteny is an alternative evolutionary pathway very different from macrosyntenic conservation. Surprisingly, mesosynteny was not found in all fungal groups. Instead, mesosynteny appears to be restricted to filamentous Ascomycetes and was most striking between species in the Dothideomycetes.
Conclusions - The existence of mesosynteny between relatively distantly related Ascomycetes could be explained by a high frequency of chromosomal inversions, but translocations must be extremely rare. The mechanism for this phenomenon is not known, but presumably involves generation of frequent inversions during meiosis.
|
|
Scooped by
Kamoun Lab @ TSL
July 25, 2011 11:57 AM
|
The fungus Mycosphaerella graminicola has been a pathogen of wheat since host domestication 10,000–12,000 years ago in the Fertile Crescent. The wheat-infecting lineage emerged from closely related Mycosphaerella pathogens infecting wild grasses. We use a comparative genomics approach to assess how the process of host specialization affected the genome structure of M. graminicola since divergence from the closest known progenitor species named M. graminicola S1. The genome of S1 was obtained by Illumina sequencing resulting in a 35 Mb draft genome sequence of 32X. Assembled contigs were aligned to the previously sequenced M. graminicola genome. The alignment covered >90% of the non-repetitive portion of the M. graminicola genome with an average divergence of 7%. The sequenced M. graminicola strain is known to harbor thirteen essential chromosomes plus eight dispensable chromosomes. We found evidence that structural rearrangements significantly affected the dispensable chromosomes while the essential chromosomes were syntenic. At the nucleotide level, the essential and dispensable chromosomes have evolved differently. The average synonymous substitution rate in dispensable chromosomes is considerably lower than in essential chromosomes, whereas the average non-synonymous substitution rate is three times higher. Differences in molecular evolution can be related to different transmission and recombination patterns, as well as to differences in effective population sizes of essential and dispensable chromosomes. In order to identify genes potentially involved in host specialization or speciation, we calculated ratios of synonymous and non-synonymous substitution rates in the >9,500 aligned protein coding genes. The genes are generally under strong purifying selection. We identified 43 candidate genes showing evidence of positive selection, one encoding a potential pathogen effector protein. We conclude that divergence of these pathogens was accompanied by structural rearrangements in the small dispensable chromosomes, while footprints of positive selection were present in only a small number of protein coding genes.
|
|
|
Rescooped by
Kamoun Lab @ TSL
from TAL effector science
September 29, 2011 9:55 AM
|
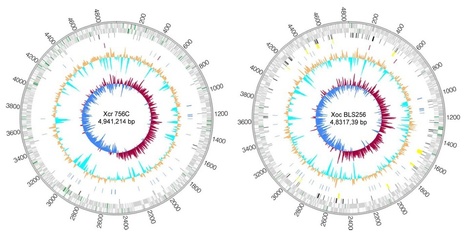
Xanthomonas is a large genus of bacteria that collectively cause disease on more than 300 plant species. The broad host range of the genus contrasts with stringent host and tissue specificity for individual species and pathovars. Whole-genome sequences of Xanthomonas campestris pv. raphani strain 756C and X. oryzae pv. oryzicola strain BLS256, pathogens that infect the mesophyll tissue of the leading models for plant biology, Arabidopsis thaliana and rice, respectively, were determined and provided insight into the genetic determinants of host and tissue specificity. Comparisons were made with genomes of closely related strains that infect the vascular tissue of the same hosts and across a larger collection of complete Xanthomonas genomes. The results suggest a model in which complex sets of adaptations at the level of gene content account for host specificity and subtler adaptations at the level of amino acid or noncoding regulatory nucleotide sequence determine tissue specificity.
Via dromius
|
|
Scooped by
Kamoun Lab @ TSL
September 16, 2011 5:22 PM
|
Emerging plant pathogens have largely been a consequence of the movement of pathogens to new geographic regions. Another documented mechanism for the emergence of plant pathogens is hybridization between individuals of different species or subspecies, which may allow rapid evolution and adaptation to new hosts or environments. Hybrid plant pathogens have traditionally been difficult to detect or confirm, but the increasing ease of cloning and sequencing PCR products now makes the identification of species that consistently have genes or alleles with phylogenetically divergent origins relatively straightforward. We investigated the genetic origin of Phytophthora andina, an increasingly common pathogen of Andean crops Solanum betaceum, S. muricatum, S. quitoense, and several wild Solanum spp. It has been hypothesized that P. andina is a hybrid between the potato late blight pathogen P. infestans and another Phytophthora species. We tested this hypothesis by cloning four nuclear loci to obtain haplotypes and using these loci to infer the phylogenetic relationships of P. andina to P. infestans and other related species. Sequencing of cloned PCR products in every case revealed two distinct haplotypes for each locus in P. andina, such that each isolate had one allele derived from a P. infestans parent and a second divergent allele derived from an unknown species that is closely related but distinct from P. infestans, P. mirabilis, and P. ipomoeae. To the best of our knowledge, the unknown parent has not yet been collected. We also observed sequence polymorphism among P. andina isolates at three of the four loci, many of which segregate between previously described P. andina clonal lineages. These results provide strong support that P. andina emerged via hybridization between P. infestans and another unknown Phytophthora species also belonging to Phytophthora clade 1c.
|
|
Scooped by
Kamoun Lab @ TSL
September 8, 2011 10:51 AM
|
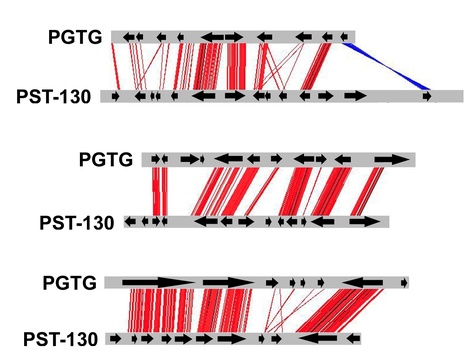
Background - The wheat stripe rust fungus (Puccinia striiformis f. sp. tritici, PST) is responsible for significant yield losses in wheat production worldwide. In spite of its economic importance, the PST genomic sequence is not currently available. Fortunately Next Generation Sequencing (NGS) has radically improved sequencing speed and efficiency with a great reduction in costs compared to traditional sequencing technologies. We used Illumina sequencing to rapidly access the genomic sequence of the highly virulent PST race 130 (PST-130). Methodology/Principal Findings - We obtained nearly 80 million high quality paired-end reads (>50x coverage) that were assembled into 29,178 contigs (64.8 Mb), which provide an estimated coverage of at least 88% of the PST genes and are available through GenBank. Extensive micro-synteny with the Puccinia graminis f. sp. tritici (PGTG) genome and high sequence similarity with annotated PGTG genes support the quality of the PST-130 contigs. We characterized the transposable elements present in the PST-130 contigs and using an ab initio gene prediction program we identified and tentatively annotated 22,815 putative coding sequences. We provide examples on the use of comparative approaches to improve gene annotation for both PST and PGTG and to identify candidate effectors. Finally, the assembled contigs provided an inventory of PST repetitive elements, which were annotated and deposited in Repbase. Conclusions/Significance - The assembly of the PST-130 genome and the predicted proteins provide useful resources to rapidly identify and clone PST genes and their regulatory regions. Although the automatic gene prediction has limitations, we show that a comparative genomics approach using multiple rust species can greatly improve the quality of gene annotation in these species. The PST-130 sequence will also be useful for comparative studies within PST as more races are sequenced. This study illustrates the power of NGS for rapid and efficient access to genomic sequence in non-model organisms.
|
|
Scooped by
Kamoun Lab @ TSL
August 30, 2011 5:47 PM
|
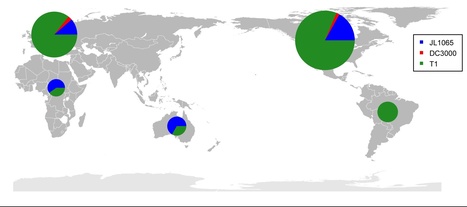
Recently, genome sequencing of many isolates of genetically monomorphic bacterial human pathogens has given new insights into pathogen microevolution and phylogeography. Here, we report a genome-based micro-evolutionary study of a bacterial plant pathogen, Pseudomonas syringae pv. tomato. Only 267 mutations were identified between five sequenced isolates in 3,543,009 nt of analyzed genome sequence, which suggests a recent evolutionary origin of this pathogen. Further analysis with genome-derived markers of 89 world-wide isolates showed that several genotypes exist in North America and in Europe indicating frequent pathogen movement between these world regions. Genome-derived markers and molecular analyses of key pathogen loci important for virulence and motility both suggest ongoing adaptation to the tomato host. A mutational hotspot was found in the type III-secreted effector gene hopM1. These mutations abolish the cell death triggering activity of the full-length protein indicating strong selection for loss of function of this effector, which was previously considered a virulence factor. Two non-synonymous mutations in the flagellin-encoding gene fliC allowed identifying a new microbe associated molecular pattern (MAMP) in a region distinct from the known MAMP flg22. Interestingly, the ancestral allele of this MAMP induces a stronger tomato immune response than the derived alleles. The ancestral allele has largely disappeared from today's Pto populations suggesting that flagellin-triggered immunity limits pathogen fitness even in highly virulent pathogens. An additional non-synonymous mutation was identified in flg22 in South American isolates. Therefore, MAMPs are more variable than expected differing even between otherwise almost identical isolates of the same pathogen strain.
|
|
Scooped by
Kamoun Lab @ TSL
August 12, 2011 1:01 AM
|
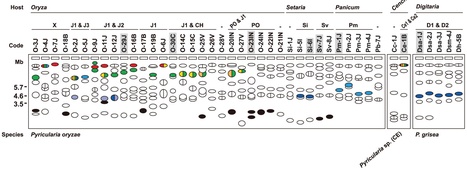
Magnaporthe oryzae is the causal agent of rice blast disease, a devastating problem worldwide. This fungus has caused breakdown of resistance conferred by newly developed commercial cultivars. To address how the rice blast fungus adapts itself to new resistance genes so quickly, we examined chromosomal locations of AVR-Pita, a subtelomeric gene family corresponding to the Pita resistance gene, in various isolates of M. oryzae (including wheat and millet pathogens) and its related species. We found that AVR-Pita (AVR-Pita1 and AVR-Pita2) is highly variable in its genome location, occurring in chromosomes 1, 3, 4, 5, 6, 7, and supernumerary chromosomes, particularly in rice-infecting isolates. When expressed in M. oryzae, most of the AVR-Pita homologs could elicit Pita-mediated resistance, even those from non-rice isolates. AVR-Pita was flanked by a retrotransposon, which presumably contributed to its multiple translocation across the genome. On the other hand, family member AVR-Pita3, which lacks avirulence activity, was stably located on chromosome 7 in a vast majority of isolates. These results suggest that the diversification in genome location of AVR-Pita in the rice isolates is a consequence of recognition by Pita in rice. We propose a model that the multiple translocation of AVR-Pita may be associated with its frequent loss and recovery mediated by its transfer among individuals in asexual populations. This model implies that the high mobility of AVR-Pita is a key mechanism accounting for the rapid adaptation toward Pita. Dynamic adaptation of some fungal plant pathogens may be achieved by deletion and recovery of avirulence genes using a population as a unit of adaptation.
|
|
Scooped by
Kamoun Lab @ TSL
August 5, 2011 4:39 AM
|
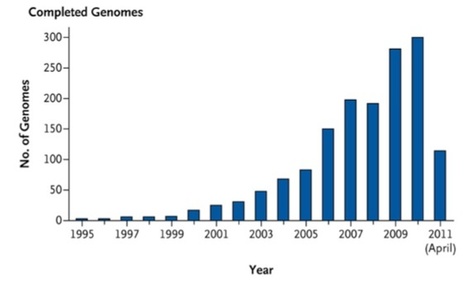
The pace of technical advancement in microbial genomics has been breathtaking. Since 1995, when the first complete genome sequence of a free-living organism, Haemophilus influenzae, was published, 1554 complete bacterial genome sequences (the majority of which are from pathogens) and 112 complete archaeal genome sequences have been determined, and more than 4800 and 90, respectively, are in progress. A total of 41 complete eukaryotic genome sequences have been determined (19 from fungi), and more than 1100 are in progress. Complete reference genome sequences are available for 2675 viral species, and for some of these species, a large number of strains have been completely sequenced. Nearly 40,000 strains of influenza virus and more than 300,000 strains of human immunodeficiency virus (HIV) type 1 have been partially sequenced. However, the selection of microbes and viruses for genome sequencing is heavily biased toward the tiny minority that are amenable to cultivation in the laboratory, numerically dominant in particular habitats of interest (e.g., the human body), and associated with disease.
|
|
Scooped by
Kamoun Lab @ TSL
July 28, 2011 10:41 AM
|
Rust fungi are some of the most devastating pathogens of crop plants. They are obligate biotrophs, which extract nutrients only from living plant tissues and cannot grow apart from their hosts. Their lifestyle has slowed the dissection of molecular mechanisms underlying host invasion and avoidance or suppression of plant innate immunity. We sequenced the 101-Mb genome of Melampsora larici-populina, the causal agent of poplar leaf rust, and the 89-Mb genome of Puccinia graminis f. sp. tritici, the causal agent of wheat and barley stem rust. We then compared the 16,399 predicted proteins of M. larici-populina with the 17,773 predicted proteins of P. graminis f. sp tritici. Genomic features related to their obligate biotrophic lifestyle include expanded lineage-specific gene families, a large repertoire of effector-like small secreted proteins, impaired nitrogen and sulfur assimilation pathways, and expanded families of amino acid and oligopeptide membrane transporters. The dramatic up-regulation of transcripts coding for small secreted proteins, secreted hydrolytic enzymes, and transporters in planta suggests that they play a role in host infection and nutrient acquisition. Some of these genomic hallmarks are mirrored in the genomes of other microbial eukaryotes that have independently evolved to infect plants, indicating convergent adaptation to a biotrophic existence inside plant cells.
|
|
Scooped by
Kamoun Lab @ TSL
July 25, 2011 6:19 AM
|
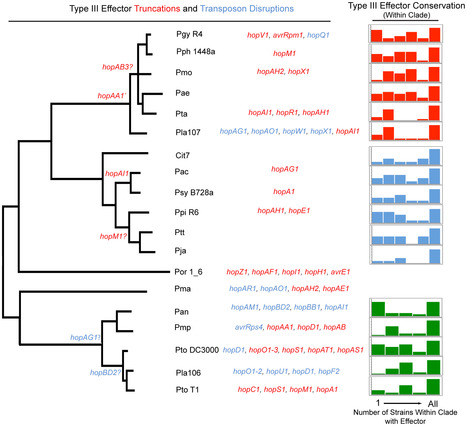
Closely related pathogens may differ dramatically in host range, but the molecular, genetic, and evolutionary basis for these differences remains unclear. In many Gram- negative bacteria, including the phytopathogen Pseudomonas syringae, type III effectors (TTEs) are essential for pathogenicity, instrumental in structuring host range, and exhibit wide diversity between strains. To capture the dynamic nature of virulence gene repertoires across P. syringae, we screened 11 diverse strains for novel TTE families and coupled this nearly saturating screen with the sequencing and assembly of 14 phylogenetically diverse isolates from a broad collection of diseased host plants. TTE repertoires vary dramatically in size and content across all P. syringae clades; surprisingly few TTEs are conserved and present in all strains. Those that are likely provide basal requirements for pathogenicity. We demonstrate that functional divergence within one conserved locus, hopM1, leads to dramatic differences in pathogenicity, and we demonstrate that phylogenetics-informed mutagenesis can be used to identify functionally critical residues of TTEs. The dynamism of the TTE repertoire is mirrored by diversity in pathways affecting the synthesis of secreted phytotoxins, highlighting the likely role of both types of virulence factors in determination of host range. We used these 14 draft genome sequences, plus five additional genome sequences previously reported, to identify the core genome for P. syringae and we compared this core to that of two closely related non-pathogenic pseudomonad species. These data revealed the recent acquisition of a 1 Mb megaplasmid by a sub-clade of cucumber pathogens. This megaplasmid encodes a type IV secretion system and a diverse set of unknown proteins, which dramatically increases both the genomic content of these strains and the pan-genome of the species.
|
|
Scooped by
Kamoun Lab @ TSL
July 25, 2011 10:15 AM
|
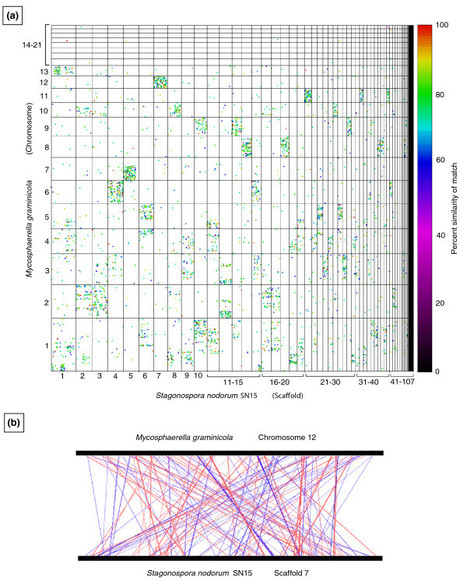
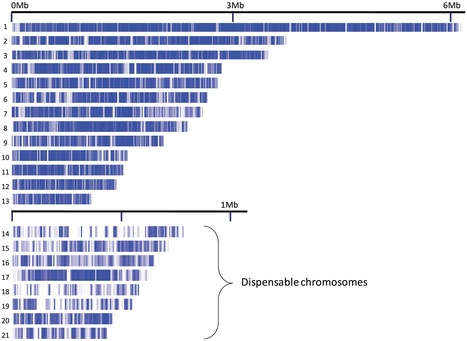
The plant-pathogenic fungus Mycosphaerella graminicola (asexual stage: Septoria tritici) causes septoria tritici blotch, a disease that greatly reduces the yield and quality of wheat. This disease is economically important in most wheat-growing areas worldwide and threatens global food production. Control of the disease has been hampered by a limited understanding of the genetic and biochemical bases of pathogenicity, including mechanisms of infection and of resistance in the host. Unlike most other plant pathogens, M. graminicola has a long latent period during which it evades host defenses. Although this type of stealth pathogenicity occurs commonly in Mycosphaerella and other Dothideomycetes, the largest class of plant-pathogenic fungi, its genetic basis is not known. To address this problem, the genome of M. graminicola was sequenced completely. The finished genome contains 21 chromosomes, eight of which could be lost with no visible effect on the fungus and thus are dispensable. This eight-chromosome dispensome is dynamic in field and progeny isolates, is different from the core genome in gene and repeat content, and appears to have originated by ancient horizontal transfer from an unknown donor. Synteny plots of the M. graminicola chromosomes versus those of the only other sequenced Dothideomycete, Stagonospora nodorum, revealed conservation of gene content but not order or orientation, suggesting a high rate of intra-chromosomal rearrangement in one or both species. This observed “mesosynteny” is very different from synteny seen between other organisms. A surprising feature of the M. graminicola genome compared to other sequenced plant pathogens was that it contained very few genes for enzymes that break down plant cell walls, which was more similar to endophytes than to pathogens. The stealth pathogenesis of M. graminicola probably involves degradation of proteins rather than carbohydrates to evade host defenses during the biotrophic stage of infection and may have evolved from endophytic ancestors.
|
|
Scooped by
Kamoun Lab @ TSL
July 25, 2011 10:13 AM
|
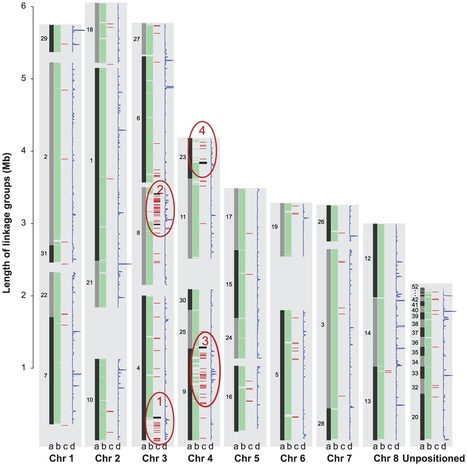
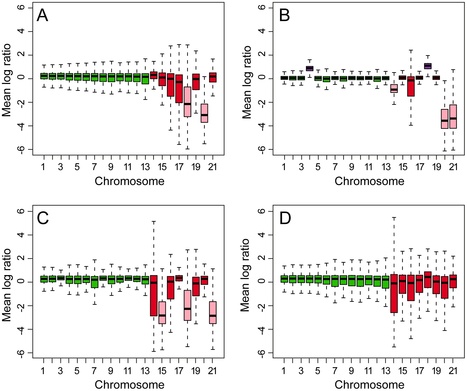
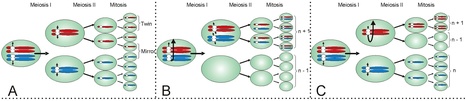
Meiosis in the haploid plant-pathogenic fungus Mycosphaerella graminicola results in eight ascospores due to a mitotic division following the two meiotic divisions. The transient diploid phase allows for recombination among homologous chromosomes. However, some chromosomes of M. graminicola lack homologs and do not pair during meiosis. Because these chromosomes are not present universally in the genome of the organism they can be considered to be dispensable. To analyze the meiotic transmission of unequal chromosome numbers, two segregating populations were generated by crossing genetically unrelated parent isolates originating from Algeria and The Netherlands that had pathogenicity towards durum or bread wheat, respectively. Detailed genetic analyses of these progenies using high-density mapping (1793 DArT, 258 AFLP and 25 SSR markers) and graphical genotyping revealed that M. graminicola has up to eight dispensable chromosomes, the highest number reported in filamentous fungi. These chromosomes vary from 0.39 to 0.77 Mb in size, and represent up to 38% of the chromosomal complement. Chromosome numbers among progeny isolates varied widely, with some progeny missing up to three chromosomes, while other strains were disomic for one or more chromosomes. Between 15–20% of the progeny isolates lacked one or more chromosomes that were present in both parents. The two high-density maps showed no recombination of dispensable chromosomes and hence, their meiotic processing may require distributive disjunction, a phenomenon that is rarely observed in fungi. The maps also enabled the identification of individual twin isolates from a single ascus that shared the same missing or doubled chromosomes indicating that the chromosomal polymorphisms were mitotically stable and originated from nondisjunction during the second division and, less frequently, during the first division of fungal meiosis. High genome plasticity could be among the strategies enabling this versatile pathogen to quickly overcome adverse biotic and abiotic conditions in wheat fields.
|