Your new post is loading...
Your new post is loading...

|
Scooped by
Kamoun Lab @ TSL
August 27, 2017 5:25 PM
|
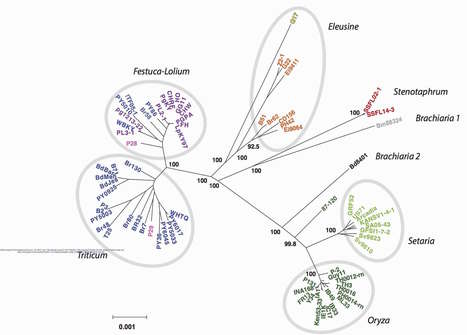
The plant immune system is innate, encoded in the germline. Using it efficiently, plants are capable of recognizing a diverse range of rapidly evolving pathogens. A recently described phenomenon shows that plant immune receptors are able to recognize pathogen effectors through the acquisition of exogenous protein domains from other plant genes. We showed that plant immune receptors with integrated domains are distributed unevenly across their phylogeny in grasses. Using phylogenetic analysis, we uncovered a major integration clade, whose members underwent repeated independent integration events producing diverse fusions. This clade is ancestral in grasses with members often found on syntenic chromosomes. Analyses of these fusion events revealed that homologous receptors can be fused to diverse domains. Furthermore, we discovered a 43 amino acids long motif that was associated with this dominant integration clade and was located immediately upstream of the fusion site. Sequence analysis revealed that DNA transposition and/or ectopic recombination are the most likely mechanisms of NLR-ID formation. The identification of this subclass of plant immune receptors that is naturally adapted to new domain integration will inform biotechnological approaches for generating synthetic receptors with novel pathogen baits.
|
|
Scooped by
Kamoun Lab @ TSL
August 25, 2017 8:21 AM
|
The rice blast fungus Magnaporthe oryzae (syn. Pyricularia oryzae) causes the most damaging rice disease. Yet, little is known about the genetic makeup of this important system for plant pathology. Using whole genome resequencing of a worldwide collection of isolates, we identified four major M. oryzae lineages mainly associated with Japonica or Indica rice, including one pandemic lineage on each rice subspecies. Tip-dating calibration indicated that M. oryzae lineages separated about a millenium ago, much later than the initial domestication of rice. The major lineage endemic to Southeast continental Asia displayed signatures of sexual recombination and evidence for having received DNA from multiple lineages. Tests of weak selection revealed that the pandemic spread of clonal lineages entailed an evolutionary 'cost', in terms of an accumulation of deleterious mutations. Our work reveals the coexistence of multiple endemic and pandemic lineages with contrasting population and genetic characteristics within a widely distributed pathogen.
|
|
Scooped by
Kamoun Lab @ TSL
August 4, 2017 8:35 AM
|
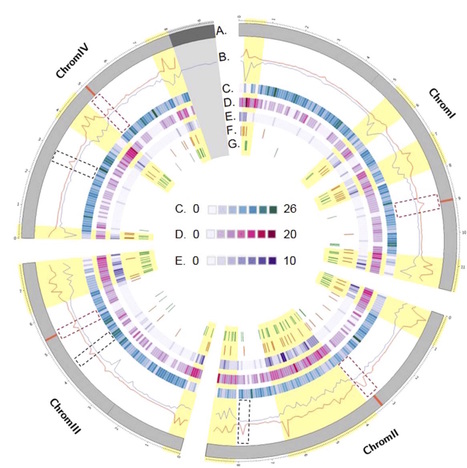
Differences in gene content are a significant source of variability within species and have an impact on phenotypic traits. However, little is known about the mechanisms responsible for the most recent gene gains and losses. We screened the genomes of 123 worldwide isolates of the major pathogen of wheat Zymoseptoria tritici for robust evidence of gene copy number variation. Based on orthology relationships in three closely related fungi, we identified 599 gene gains and 1,024 gene losses that have not yet reached fixation within the focal species. Our analyses of gene gains and losses segregating in populations showed that gene copy number variation arose preferentially in subtelomeres and in proximity to transposable elements. Recently lost genes were enriched in virulence factors and secondary metabolite gene clusters. In contrast, recently gained genes encoded mostly secreted protein lacking a conserved domain. We analyzed the frequency spectrum at loci segregating a gene presence–absence polymorphism in four worldwide populations. Recent gene losses showed a significant excess in low-frequency variants compared with genome-wide single nucleotide polymorphism, which is indicative of strong negative selection against gene losses. Recent gene gains were either under weak negative selection or neutral. We found evidence for strong divergent selection among populations at individual loci segregating a gene presence–absence polymorphism. Hence, gene gains and losses likely contributed to local adaptation. Our study shows that microbial eukaryotes harbor extensive copy number variation within populations and that functional differences among recently gained and lost genes led to distinct evolutionary trajectories.
|
|
Scooped by
Kamoun Lab @ TSL
August 3, 2017 8:00 AM
|
Transcription activator-like effectors (TALEs) are virulence factors of plant-pathogenic Xanthomonas spp. that function as gene activators inside plant host cells. AnnoTALE is a suite of applications for identifying and analysing TALEs in Xanthomonas genomes, for clustering TALEs into classes by their RVD sequences, for assigning novel TALEs to existing classes, for proposing TALE names using a unified nomenclature, and for predicting targets of individual TALEs and TALE classes. AnnoTALE is available as a JavaFX-based stand-alone application with graphical user interface for interactive analysis sessions. In addition, we provide a command line application that may be integrated into other pipelines. Both use identical code for the actual analysis, ensuring consistent results between both versions. If you use AnnoTALE, please cite: Jan Grau, Maik Reschke, Annett Erkes, Jana Streubel, Richard D. Morgan, Geoffrey G. Wilson, Ralf Koebnik and Jens Boch. AnnoTALE: bioinformatics tools for identification, annotation, and nomenclature of TALEs from Xanthomonas genomic sequences. Scientific Reports 6:21077, DOI: 10.1038/srep21077, 2016.
|
|
Scooped by
Kamoun Lab @ TSL
August 3, 2017 1:16 AM
|
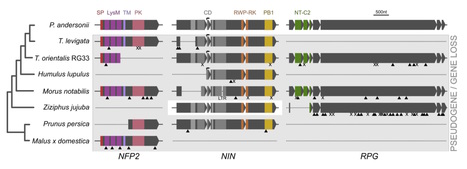
Rhizobium nitrogen-fixing nodules are a well-known trait of legumes, but nodules also occur in other plant lineages either with rhizobium or the actinomycete Frankia as microsymbiont. The widely accepted hypothesis is that nodulation evolved independently multiple times, with only a few losses. However, insight in the evolutionary trajectory of nodulation is lacking. We conducted comparative studies using Parasponia (Cannabaceae), the only non-legume able to establish nitrogen fixing nodules with rhizobium. This revealed that Parasponia and legumes utilize a large set of orthologous symbiosis genes. Comparing genomes of Parasponia and its non-nodulating relative Trema did not reveal specific gene duplications that could explain a recent gain of nodulation in Parasponia. Rather, Trema and other non-nodulating species in the order Rosales show evidence of pseudogenization or loss of key symbiosis genes. This demonstrates that these species have lost the potential to nodulate. This finding challenges a long-standing hypothesis on evolution of nitrogen-fixing symbioses, and has profound implications for translational approaches aimed at engineering nitrogen-fixing nodules in crop plants.
|
|
Scooped by
Kamoun Lab @ TSL
July 24, 2017 7:34 PM
|
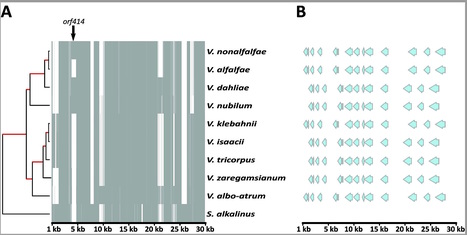
The fungal genus Verticillium contains ten species, some of which are notorious plant pathogens causing vascular wilt diseases in host plants, while others are known as saprophytes and opportunistic plant pathogens. Whereas the genome of V. dahliae, the most notorious plan pathogen of the genus, has been well characterized, evolution and speciation of other members of the genus received little attention thus far. Here, we sequenced the genomes of the nine haploid Verticillium spp. to study evolutionary trajectories of their divergence from a last common ancestor. Frequent occurrence of chromosomal rearrangement and gene family loss was identified. In addition to ~11,000 core genes that are shared among all species, only 200-600 species-specific genes occur. Intriguingly, these species-specific genes show different features than core genes.
|
|
Scooped by
Kamoun Lab @ TSL
July 18, 2017 5:00 AM
|
Sclerotinia sclerotiorum is a phytopathogenic fungus with over 400 hosts including numerous economically important cultivated species. This contrasts many economically destructive pathogens that only exhibit a single or very few hosts. Many plant pathogens exhibit a “two-speed” genome. So described because their genomes contain alternating gene rich, repeat sparse and gene poor, repeat-rich regions. In fungi, the repeat-rich regions may be subjected to a process termed repeat-induced point mutation (RIP). Both repeat activity and RIP are thought to play a significant role in evolution of secreted virulence proteins, termed effectors. We present a complete genome sequence of S. sclerotiorum generated using Single Molecule Real-Time Sequencing technology with highly accurate annotations produced using an extensive RNA sequencing data set. We identified 70 effector candidates and have highlighted their in planta expression profiles. Furthermore, we characterized the genome architecture of S. sclerotiorum in comparison to plant pathogens that exhibit “two-speed” genomes. We show that there is a significant association between positions of secreted proteins and regions with a high RIP index in S. sclerotiorum but we did not detect a correlation between secreted protein proportion and GC content. Neither did we detect a negative correlation between CDS content and secreted protein proportion across the S. sclerotiorumgenome. We conclude that S. sclerotiorum exhibits subtle signatures of enhanced mutation of secreted proteins in specific genomic compartments as a result of transposition and RIP activity. However, these signatures are not observable at the whole-genome scale.
|
|
Scooped by
Kamoun Lab @ TSL
May 11, 2017 4:13 AM
|
Click here to edit the content
|
|
Scooped by
Kamoun Lab @ TSL
May 10, 2017 9:40 AM
|
Fungal plant pathogens rapidly evolve virulence on resistant hosts through mutations in genes encoding proteins that modulate the host immune responses. The mutational spectrum likely includes chromosomal rearrangements responsible for gains or losses of entire genes. However, the mechanisms creating adaptive structural variation in fungal pathogen populations are poorly understood. We used complete genome assemblies to quantify structural variants segregating in the highly polymorphic fungal wheat pathogen Zymoseptoria tritici. The genetic basis of virulence in Z. tritici is complex, and populations harbor significant genetic variation for virulence; hence, we aimed to identify whether structural variation led to functional differences. We combined single-molecule real-time sequencing, genetic maps, and transcriptomics data to generate a fully assembled and annotated genome of the highly virulent field isolate 3D7. Comparative genomics analyses against the complete reference genome IPO323 identified large chromosomal inversions and the complete gain or loss of transposable-element clusters, explaining the extensive chromosomal-length polymorphisms found in this species. Both the 3D7 and IPO323 genomes harbored long tracts of sequences exclusive to one of the two genomes. These orphan regions contained 296 genes unique to the 3D7 genome and not previously known for this species. These orphan genes tended to be organized in clusters and showed evidence of mutational decay. Moreover, the orphan genes were enriched in genes encoding putative effectors and included a gene that is one of the most upregulated putative effector genes during wheat infection. Our study showed that this pathogen species harbored extensive chromosomal structure polymorphism that may drive the evolution of virulence.
|
|
Scooped by
Kamoun Lab @ TSL
May 10, 2017 8:32 AM
|
Recombination is a major evolutionary force, increasing genetic diversity and permitting efficient coevolution of fungal pathogen(s) with their host(s). The ascomycete Fusarium graminearum is a devastating pathogen of cereal crops, and can contaminate food and feed with harmful mycotoxins. Previous studies have suggested a high adaptive potential of this pathogen, illustrated by an increase in pathogenicity and resistance to fungicides. In this study, we provide the first detailed picture of the crossover events occurring during meiosis and discuss the role of recombination in pathogen evolution. An experimental recombinant population (n = 88) was created and genotyped using 1306 polymorphic markers obtained from restriction site-associated DNA sequencing (RAD-seq) and aligned to the reference genome. The construction of a high-density linkage map, anchoring 99% of the total length of the reference genome, allowed the identification of 1451 putative crossovers, positioned at a median resolution of 24 kb. The majority of crossovers (87.2%) occurred in a relatively small portion of the genome (30%). All chromosomes demonstrated recombination-active sections, which had a near 15-fold higher crossover rate than non-active recombinant sections. The recombination rate showed a strong positive correlation with nucleotide diversity, and recombination-active regions were enriched for genes with a putative role in host–pathogen interaction, as well as putative diversifying genes. Our results confirm the preliminary analysis observed in other F. graminearum strains and suggest a conserved ‘two-speed’ recombination landscape. The consequences with regard to the evolutionary potential of this major fungal pathogen are also discussed.
|
|
Rescooped by
Kamoun Lab @ TSL
from Publications
April 5, 2017 8:23 AM
|
Background. In February 2016, a new fungal disease was spotted in wheat fields across eight districts in Bangladesh. The epidemic spread to an estimated 15,000 hectares, about 16 % of the cultivated wheat area in Bangladesh, with yield losses reaching up to 100 %. Within weeks of the onset of the epidemic, we performed transcriptome sequencing of symptomatic leaf samples collected directly from Bangladeshi fields.
Results. Reinoculation of seedlings with strains isolated from infected wheat grains showed wheat blast symptoms on leaves of wheat but not rice. Our phylogenomic and population genomic analyses revealed that the wheat blast outbreak in Bangladesh was most likely caused by a wheat-infecting South American lineage of the blast fungus Magnaporthe oryzae.
Conclusion. Our findings suggest that genomic surveillance can be rapidly applied to monitor plant disease outbreaks and provide valuable information regarding the identity and origin of the infectious agent.
|
|
Scooped by
Kamoun Lab @ TSL
March 11, 2017 3:25 PM
|
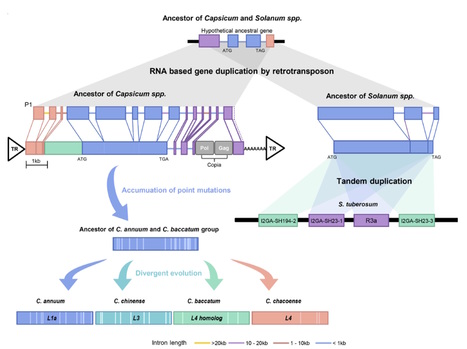
Transposable elements (TEs) provide major evolutionary forces leading to new genome structure and species diversification. However, the role of TEs in the expansion of disease resistance gene families has been unexplored in plants. Here, we report high-quality de novo genomes for two peppers (Capsicum baccatum and C. chinense) and an improved reference genome (C. annuum). Dynamic genome rearrangements involving translocations among chromosome 3, 5 and 9 were detected in comparison between C. baccatum and the two other peppers. The amplification of athila LTR-retrotransposons, members of the gypsy superfamily, led to genome expansion in C. baccatum. In-depth genome-wide comparison of genes and repeats unveiled that the copy numbers of NLRs were greatly increased by LTR-retrotransposon-mediated retroduplication. Moreover, retroduplicated NLRs exhibited great abundance across the angiosperms, with most cases lineage-specific and thus recent events. Our study revealed that retroduplication has played key roles in the emergence of new disease-resistance genes in plants.
|
|
Scooped by
Kamoun Lab @ TSL
February 15, 2017 5:25 AM
|
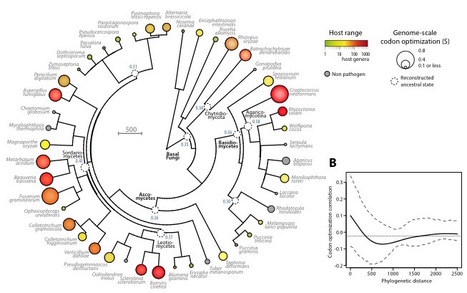
The range of hosts that parasites can infect is a key determinant of the emergence and spread of disease. Yet the impact of host range variation on the evolution of parasite genomes remains unknown. Here, we show that codon optimization underlies genome adaptation in broad host range parasite. We found that the longer proteins encoded by broad host range fungi likely increase natural selection on codon optimization in these species. Accordingly, codon optimization correlates with host range across the fungal kingdom. At the species level, biased patterns of synonymous substitutions underpin increased codon optimization in a generalist but not a specialist fungal pathogen. Virulence genes were consistently enriched in highly codon-optimized genes of generalist but not specialist species. We conclude that codon optimization is related to the capacity of parasites to colonize multiple hosts. Our results link genome evolution and translational regulation to the long term persistence of generalist parasitism.
|
|
|
Scooped by
Kamoun Lab @ TSL
August 25, 2017 11:26 AM
|
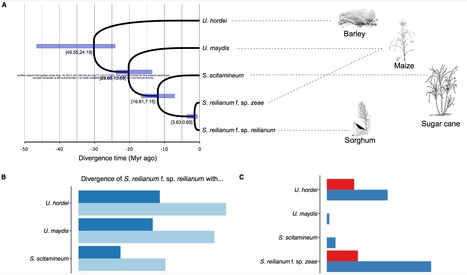
Plants and fungi display a broad range of interactions within natural and agricultural ecosystems ranging from symbiosis to parasitism. Pathogenic interactions are governed by secreted fungal effector proteins, which are thought to coevolve with their host targets. Biotrophic smut fungi which belong to the division of Basidiomycota are well-suited to investigate the evolution of plant pathogens, because several quality draft genomes and genetic tools are available for these species. Here, we used the genomes of Sporisorium reilianum f. sp. zeae and S. reilianum f. sp. reilianum, two closely related formae speciales infecting maize and sorghum, respectively, together with the genomes of Ustilago hordei, U. maydis and S. scitamineum to identify effector genes showing signs of positive selection. The largest numbers of such genes were identified in the two pathovariants of S. reilianum and between paralogues in U. hordei, where many belong to families showing species-specific expansions. Next, we assessed the contribution to virulence of candidate effector genes in S. reilianum f. sp. zeae by deletion of individual genes in a solopathogenic strain. While eight of nine deletions mutants were unaffected in virulence, one mutant had lost virulence. This shows that despite the relatively recent divergent time of the two formae speciales, a signature of positive selection in candidate effector genes in S. reilianum is a poor indicator for the identification of genes with virulence functions.
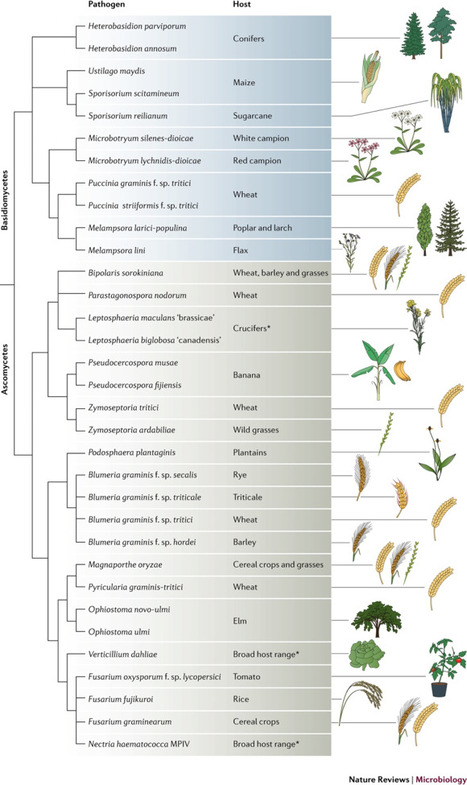
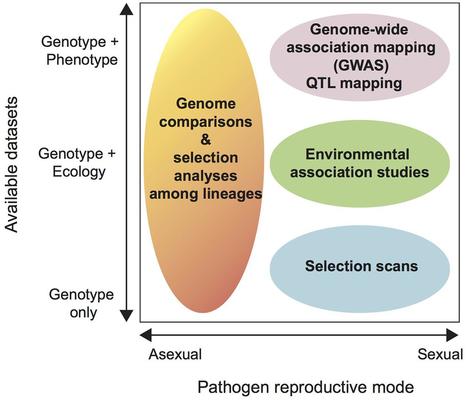
The fungal kingdom comprises some of the most devastating plant pathogens. Sequencing the genomes of fungal pathogens has shown a remarkable variability in genome size and architecture. Population genomic data enable us to understand the mechanisms and the history of changes in genome size and adaptive evolution in plant pathogens. Although transposable elements predominantly have negative effects on their host, fungal pathogens provide prominent examples of advantageous associations between rapidly evolving transposable elements and virulence genes that cause variation in virulence phenotypes. By providing homogeneous environments at large regional scales, managed ecosystems, such as modern agriculture, can be conducive for the rapid evolution and dispersal of pathogens. In this Review, we summarize key examples from fungal plant pathogen genomics and discuss evolutionary processes in pathogenic fungi in the context of molecular evolution, population genomics and agriculture.
Via Ronny Kellner
|
|
Scooped by
Kamoun Lab @ TSL
August 3, 2017 8:03 AM
|
Transcription activator-like effectors (TALEs) are secreted by plant–pathogenic Xanthomonas bacteria into plant cells where they act as transcriptional activators and, hence, are major drivers in reprogramming the plant for the benefit of the pathogen. TALEs possess a highly repetitive DNA-binding domain of typically 34 amino acid (AA) tandem repeats, where AA 12 and 13, termed repeat variable di-residue (RVD), determine target specificity. Different Xanthomonas strains possess different repertoires of TALEs. Here, we study the evolution of TALEs from the level of RVDs determining target specificity down to the level of DNA sequence with focus on rice-pathogenic Xanthomonas oryzae pv. oryzae(Xoo) and Xanthomonas oryzae pv. oryzicola (Xoc) strains. We observe that codon pairs coding for individual RVDs are conserved to a similar degree as the flanking repeat sequence. We find strong indications that TALEs may evolve 1) by base substitutions in codon pairs coding for RVDs, 2) by recombination of N-terminal or C-terminal regions of existing TALEs, or 3) by deletion of individual TALE repeats, and we propose possible mechanisms. We find indications that the reassortment of TALE genes in clusters is mediated by an integron-like mechanism in Xoc. We finally study the effect of the presence/absence and evolutionary modifications of TALEs on transcriptional activation of putative target genes in rice, and find that even single RVD swaps may lead to considerable differences in activation. This correlation allowed a refined prediction of TALE targets, which is the crucial step to decipher their virulence activity.
|
|
Scooped by
Kamoun Lab @ TSL
August 3, 2017 1:28 AM
|
Cotton bacterial blight (CBB), an important disease of (Gossypium hirsutum) in the early 20th century, had been controlled by resistant germplasm for over half a century. Recently, CBB re-emerged as an agronomic problem in the United States. Here, we report analysis of cotton variety planting statistics that indicate a steady increase in the percentage of susceptible cotton varieties grown each year since 2009. Phylogenetic analysis revealed that strains from the current outbreak cluster with race 18 Xanthomonas citri pv. malvacearum (Xcm) strains. Illumina based draft genomes were generated for thirteen Xcm isolates and analyzed along with 4 previously published Xcm genomes. These genomes encode 24 conserved and nine variable type three effectors. Strains in the race 18 clade contain 3 to 5 more effectors than other Xcm strains. SMRT sequencing of two geographically and temporally diverse strains of Xcm yielded circular chromosomes and accompanying plasmids. These genomes encode eight and thirteen distinct transcription activator-like effector genes. RNA-sequencing revealed 52 genes induced within two cotton cultivars by both tested Xcm strains. This gene list includes a homeologous pair of genes, with homology to the known susceptibility gene, MLO. In contrast, the two strains of Xcm induce different clade III SWEET sugar transporters. Subsequent genome wide analysis revealed patterns in the overall expression of homeologous gene pairs in cotton after inoculation by Xcm. These data reveal host-pathogen specificity at the genetic level and strategies for future development of resistant cultivars.
|
|
Rescooped by
Kamoun Lab @ TSL
from Plant pathogenic fungi
August 2, 2017 4:37 AM
|
The ability of an organism to replicate and segregate its genome with high fidelity is vital to its survival and for the production of future generations. Errors in either of these steps (replication or segregation) can lead to a change in ploidy or chromosome number. While these drastic genome changes can be detrimental to the organism, resulting in decreased fitness, they can also provide increased fitness during periods of stress. A change in ploidy or chromosome number can fundamentally change how a cell senses and responds to its environment. Here, we discuss current ideas in fungal biology that illuminate how eukaryotic genome size variation can impact the organism at a cellular and evolutionary level. One of the most fascinating observations from the past 2 decades of research is that some fungi have evolved the ability to tolerate large genome size changes and generate vast genomic heterogeneity without undergoing canonical meiosis.
Via Niklaus Grunwald, Steve Marek
|
|
Scooped by
Kamoun Lab @ TSL
July 23, 2017 9:45 AM
|
Delineating species and epidemic lineages in fungal plant pathogens is critical to our understanding of disease emergence and the structure of fungal biodiversity, and also informs international regulatory decisions. Pyricularia oryzae (syn. Magnaporthe oryzae) is a multi-host pathogen that infects multiple grasses and cereals, is responsible for the most damaging rice disease (rice blast), and of growing concern due to the recent introduction of wheat blast to Bangladesh from South America. However, the genetic structure and evolutionary history of M. oryzae, including the possible existence of cryptic phylogenetic species, remain poorly defined. Here, we use whole-genome sequence information for 76 M. oryzae isolates sampled from 12 grass and cereal genera to infer the population structure of M. oryzae, and to reassess the species status of wheat-infecting populations of the fungus. Species recognition based on genealogical concordance, using published data or extracting previously-used loci from genome assemblies, failed to confirm a prior assignment of wheat blast isolates to a new species (Pyricularia graminis tritici). Inference of population subdivisions revealed multiple divergent lineages within M. oryzae, each preferentially associated with one host genus, suggesting incipient speciation following host shift or host range expansion. Analyses of gene flow, taking into account the possibility of incomplete lineage sorting, revealed that genetic exchanges have contributed to the makeup of multiple lineages within M. oryzae. These findings provide greater understanding of the eco-evolutionary factors that underlie the diversification of M. oryzae and highlight the practicality of genomic data for epidemiological surveillance in this important multi-host pathogen.
|
|
Scooped by
Kamoun Lab @ TSL
May 13, 2017 6:37 AM
|
Background. Plant-pathogenic oomycetes are responsible for economically important losses in crops worldwide. Phytophthora palmivora, a tropical relative of the potato late blight pathogen, causes rotting diseases in many tropical crops including papaya, cocoa, oil palm, black pepper, rubber, coconut, durian, mango, cassava and citrus. Transcriptomics have helped to identify repertoires of host-translocated microbial effector proteins which counteract defenses and reprogram the host in support of infection. As such, these studies have helped in understanding how pathogens cause diseases. Despite the importance of P. palmivora diseases, genetic resources to allow for disease resistance breeding and identification of microbial effectors are scarce. Results. We employed the model plant Nicotiana benthamiana to study the P. palmivora root infections at the cellular and molecular levels. Time-resolved dual transcriptomics revealed different pathogen and host transcriptome dynamics. De novo assembly of P. palmivora transcriptome and semi-automated prediction and annotation of the secretome enabled robust identification of conserved infection-promoting effectors. We show that one of them, REX3, suppresses plant secretion processes. In a survey for early transcriptionally activated plant genes we identified a N. benthamiana gene specifically induced at infected root tips that encodes a peptide with danger-associated molecular features. Conclusions. These results constitute a major advance in our understanding of P. palmivora diseases and establish extensive resources for P. palmivora pathogenomics, effector-aided resistance breeding and the generation of induced resistance to Phytophthora root infections. Furthermore, our approach to find infection-relevant secreted genes is transferable to other pathogen-host interactions and not restricted to plants.
|
|
Scooped by
Kamoun Lab @ TSL
May 10, 2017 10:31 AM
|
Sex chromosomes in plants and animals and fungal mating-type chromosomes often show exceptional genome features, with extensive suppression of homologous recombination and cytological differentiation between members of the diploid chromosome pair. Despite strong interest in the genetics of these chromosomes, their large regions of suppressed recombination often are enriched in transposable elements and therefore can be challenging to assemble. Here we show that the latest improvements of the PacBio sequencing yield assembly of the whole genome of the anther-smut fungus, Microbotryum lychnidis-dioicae (the pathogenic fungus causing anther-smut disease of Silene latifolia), into finished chromosomes or chromosome arms, even for the repeat-rich mating-type chromosomes and centromeres. Suppressed recombination of the mating-type chromosomes is revealed to span nearly 90% of their lengths, with extreme levels of rearrangements, transposable element accumulation, and differentiation between the two mating types. We observed no correlation between allelic divergence and physical position in the nonrecombining regions of the mating-type chromosomes. This may result from gene conversion or from rearrangements of ancient evolutionary strata, i.e., successive steps of suppressed recombination. Centromeres were found to be composed mainly of copia-like transposable elements and to possess specific minisatellite repeats identical between the different chromosomes. We also identified subtelomeric motifs. In addition, extensive signs of degeneration were detected in the nonrecombining regions in the form of transposable element accumulation and of hundreds of gene losses on each mating-type chromosome. Furthermore, our study highlights the potential of the latest breakthrough PacBio chemistry to resolve complex genome architectures.
|
|
Scooped by
Kamoun Lab @ TSL
May 10, 2017 9:36 AM
|
Epidemics caused by fungal plant pathogens pose a major threat to agro-ecosystems and impact global food security. High-throughput sequencing enabled major advances in understanding how pathogens cause disease on crops. Hundreds of fungal genomes are now available and analyzing these genomes highlighted the key role of effector genes in disease. Effectors are small secreted proteins that enhance infection by manipulating host metabolism. Fungal genomes carry 100s of putative effector genes, but the lack of homology among effector genes, even for closely related species, challenges evolutionary and functional analyses. Furthermore, effector genes are often found in rapidly evolving chromosome compartments which are difficult to assemble. We review how population and comparative genomics toolsets can be combined to address these challenges. We highlight studies that associated genome-scale polymorphisms with pathogen lifestyles and adaptation to different environments. We show how genome-wide association studies can be used to identify effectors and other pathogenicity-related genes underlying rapid adaptation. We also discuss how the compartmentalization of fungal genomes into core and accessory regions shapes the evolution of effector genes. We argue that an understanding of genome evolution provides important insight into the trajectory of host-pathogen co-evolution.
|
|
Scooped by
Kamoun Lab @ TSL
April 7, 2017 10:30 AM
|
|
|
Scooped by
Kamoun Lab @ TSL
March 12, 2017 3:58 AM
|
Outbreaks caused by asexual lineages of fungal and oomycete pathogens are an expanding threat to crops, wild animals and natural ecosystems (Fisher et al. 2012, Kupferschmidt 2012). However, the mechanisms underlying genome evolution and phenotypic plasticity in asexual eukaryotic microbes remain poorly understood (Seidl and Thomma 2014). Ever since the 19th century Irish famine, the oomycete Phytophthora infestans has caused recurrent outbreaks on potato and tomato crops that have been primarily caused by the successive rise and migration of pandemic asexual lineages (Cooke et al. 2012, Yoshida et al. 2013, Yoshida et al. 2014). Here, we reveal patterns of genomic and gene expression variation within a P. infestans asexual lineage by compared sibling strains belonging to the South American EC-1 clone that has dominated Andean populations since the 1990s (Forbes et al. 1997, Oyarzun et al. 1998, Delgado et al. 2013, Yoshida et al. 2013, Yoshida et al. 2014). We detected numerous examples of structural variation, nucleotide polymorphisms and gene conversion within the EC-1 clone. Remarkably, 17 genes are not expressed in one of the two EC-1 isolates despite apparent absence of sequence polymorphisms. Among these, silencing of an effector gene was associated with evasion of disease resistance conferred by a potato immune receptor. These results highlight the exceptional genetic and phenotypic plasticity that underpins host adaptation in a pandemic clonal lineage of a eukaryotic plant pathogen.
|
|
Scooped by
Kamoun Lab @ TSL
March 1, 2017 4:40 AM
|
Cercospora species have a global distribution and are best known as the causal agents of leaf spot diseases of many crops. Cercospora leaf spot (CLS) is an economically devastating disease of sugar beet caused by C. beticola. The C. beticola genome encodes 63 biosynthetic gene clusters, including the cercosporin toxin biosynthesis (CTB) cluster. Studies spanning nearly 60 years have shown that cercosporin is photo-activated, critical for disease development, and toxic to most organisms except Cercospora spp. themselves, which exhibit cercosporin auto-resistance. We show that the CTB gene cluster has experienced an unprecedented number of duplications, losses, and horizontal transfers across a spectrum of plant pathogenic fungi. Although cercosporin biosynthesis has been widely assumed to rely on the eight gene CTB cluster, our comparative genomic analysis revealed extensive gene collinearity adjacent to the established cluster in all CTB cluster-harboring species. We demonstrate that the CTB cluster is larger than previously recognized and includes the extracellular proteins fasciclin and laccase required for cercosporin biosynthesis and the final pathway enzyme that installs the unusual cercosporin methylenedioxy bridge. Additionally, the expanded cluster contains CFP, which contributes to cercosporin auto-resistance in C. beticola. Together, our results give new insight on the intricate evolution of the CTB cluster.
|