Your new post is loading...
Your new post is loading...

|
Scooped by
Kamoun Lab @ TSL
August 28, 2015 4:25 AM
|
Next-generation sequencing (NGS) technologies have increased the scalability, speed, and resolution of genomic sequencing and, thus, have revolutionized genomic studies. However, eukaryotic genome sequencing initiatives typically yield considerably fragmented genome assemblies. Here, we assessed various state-of-the-art sequencing and assembly strategies in order to produce a contiguous and complete eukaryotic genome assembly, focusing on the filamentous fungus Verticillium dahliae. Compared with Illumina-based assemblies of the V. dahliae genome, hybrid assemblies that also include PacBio-generated long reads establish superior contiguity. Intriguingly, provided that sufficient sequence depth is reached, assemblies solely based on PacBio reads outperform hybrid assemblies and even result in fully assembled chromosomes. Furthermore, the addition of optical map data allowed us to produce a gapless and complete V. dahliae genome assembly of the expected eight chromosomes from telomere to telomere. Consequently, we can now study genomic regions that were previously not assembled or poorly assembled, including regions that are populated by repetitive sequences, such as transposons, allowing us to fully appreciate an organism’s biological complexity. Our data show that a combination of PacBio-generated long reads and optical mapping can be used to generate complete and gapless assemblies of fungal genomes.
IMPORTANCE Studying whole-genome sequences has become an important aspect of biological research. The advent of next-generation sequencing (NGS) technologies has nowadays brought genomic science within reach of most research laboratories, including those that study nonmodel organisms. However, most genome sequencing initiatives typically yield (highly) fragmented genome assemblies. Nevertheless, considerable relevant information related to genome structure and evolution is likely hidden in those nonassembled regions. Here, we investigated a diverse set of strategies to obtain gapless genome assemblies, using the genome of a typical ascomycete fungus as the template. Eventually, we were able to show that a combination of PacBio-generated long reads and optical mapping yields a gapless telomere-to-telomere genome assembly, allowing in-depth genome analyses to facilitate functional studies into an organism’s biology.
|
|
Scooped by
Kamoun Lab @ TSL
August 4, 2015 9:02 AM
|
The speciation of pathogens can be driven by divergent host specialization. Specialization to a new host is possible via the acquisition of advantageous mutations fixed by positive selection. Comparative genome analyses of closely related species allows for the identification of such key substitutions via inference of genome-wide signatures of positive selection. We previously used a comparative genomics framework to identify genes that have evolved under positive selection during speciation of the prominent wheat pathogen Zymoseptoria tritici (synonym Mycosphaerella graminicola). In this study, we conducted functional analyses of four genes exhibiting strong signatures of positive selection in Z. tritici. We deleted the four genes in Z. tritici and confirm a virulence-related role of three of the four genes ΔZt80707, ΔZt89160 and ΔZt103264. The two mutants ΔZt80707 and ΔZt103264 show a significant reduction in virulence during infection of wheat; the ΔZt89160 mutant causes a hypervirulent phenotype in wheat. Mutant phenotypes of ΔZt80707, ΔZt89160 and ΔZt103264 can be restored by insertion of the wild-type genes. However, the insertion of the Zt80707 and Zt89160 orthologs from Z. pseudotritici and Z. ardabiliae do not restore wild-type levels of virulence, suggesting that positively selected substitutions in Z. tritici may relate to divergent host specialization. Interestingly, the gene Zt80707 encodes also a secretion signal that targets the protein for cell secretion. This secretion signal is however only transcribed in Z. tritici, suggesting that Z. tritici-specific substitutions relate to a new function of the protein in the extracellular space of the wheat-Z. tritici interaction. Together, the results presented here highlight that Zt80707, Zt103264 and Zt89160 represent key genes involved in virulence and host-specific disease development of Z. tritici. Our findings illustrate that evolutionary predictions provide a powerful tool for the identification of novel traits crucial for host adaptation and pathogen evolution.
|
|
Scooped by
Kamoun Lab @ TSL
July 30, 2015 5:37 AM
|
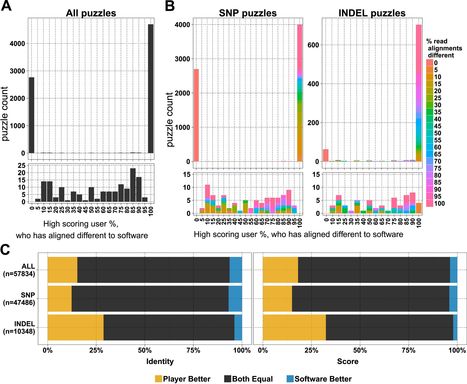
In 2013, in response to an epidemic of ash dieback disease in England the previous year, we launched a Facebook-based game called Fraxinus to enable non-scientists to contribute to genomics studies of the pathogen that causes the disease and the ash trees that are devastated by it. Over a period of 51 weeks players were able to match computational alignments of genetic sequences in 78% of cases, and to improve them in 15% of cases. We also found that most players were only transiently interested in the game, and that the majority of the work done was performed by a small group of dedicated players. Based on our experiences we have built a linear model for the length of time that contributors are likely to donate to a crowd-sourced citizen science project. This model could serve a guide for the design and implementation of future crowd-sourced citizen science initiatives. - See more at: http://elifesciences.org/content/4/e07460#.dpuf
|
|
Scooped by
Kamoun Lab @ TSL
June 22, 2015 6:34 AM
|
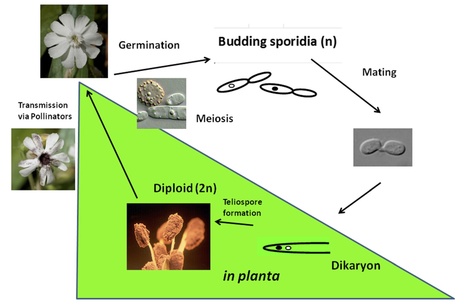
Background. The genus Microbotryum includes plant pathogenic fungi afflicting a wide variety of hosts with anther smut disease. Microbotryum lychnidis-dioicae infects Silene latifolia and replaces host pollen with fungal spores, exhibiting biotrophy and necrosis associated with altering plant development.
Results. We determined the haploid genome sequence for M. lychnidis-dioicae and analyzed whole transcriptome data from plant infections and other stages of the fungal lifecycle, revealing the inventory and expression level of genes that facilitate pathogenic growth. Compared to related fungi, an expanded number of major facilitator superfamily transporters and secretory lipases were detected; lipase gene expression was found to be altered by exposure to lipid compounds, which signaled a switch to dikaryotic, pathogenic growth. In addition, while enzymes to digest cellulose, xylan, xyloglucan, and highly substituted forms of pectin were absent, along with depletion of peroxidases and superoxide dismutases that protect the fungus from oxidative stress, the repertoire of glycosyltransferases and of enzymes that could manipulate host development has expanded. A total of 14 % of the genome was categorized as repetitive sequences. Transposable elements have accumulated in mating-type chromosomal regions and were also associated across the genome with gene clusters of small secreted proteins, which may mediate host interactions.
Conclusions. The unique absence of enzyme classes for plant cell wall degradation and maintenance of enzymes that break down components of pollen tubes and flowers provides a striking example of biotrophic host adaptation.
|
|
Scooped by
Kamoun Lab @ TSL
April 14, 2015 5:37 AM
|
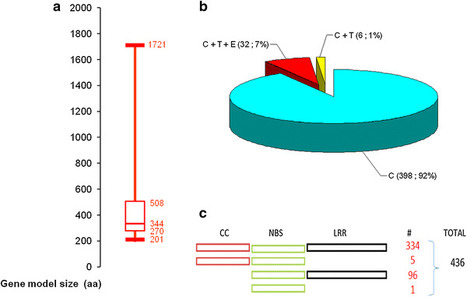
Host resistance is the most economical, effective and ecologically sustainable method of controlling diseases in crop plants. In bread wheat, despite the high number of resistance loci that have been cataloged to date, only few have been cloned, underlying the need for genomics-guided investigations capable of providing a prompt and acute knowledge on the identity of effective resistance genes that can be used in breeding programs. Proteins with a nucleotide-binding site (NBS) encoded by the major plant disease resistance (R) genes play an important role in the responses of plants to various pathogens. In this study, a comprehensive analysis of NBS-encoding genes within the whole wheat genome was performed, and the genome scale characterization of this gene family was established. From the recently published wheat genome sequence, we used a data mining and automatic prediction pipeline to identify 580 complete ORF candidate NBS-encoding genes and 1,099 partial-ORF ones. Among complete gene models, 464 were longer than 200 aa, among them 436 had less than 70 % of sequence identity to each other. This gene models set was deeply characterized. (1) First, we have analyzed domain architecture and identified, in addition to typical domain combinations, the presence of particular domains like signal peptides, zinc fingers, kinases, heavy-metal-associated and WRKY DNA-binding domains. (2) Functional and expression annotation via homology searches in protein and transcript databases, based on sufficient criteria, enabled identifying similar proteins for 60 % of the studied gene models and expression evidence for 13 % of them. (3) Shared orthologous groups were defined using NBS-domain proteins of rice and Brachypodium distachyon. (4) Finally, alignment of the 436 NBS-containing gene models to the full set of scaffolds from the IWGSC’s wheat chromosome survey sequence enabled high-stringence anchoring to chromosome arms. The distribution of the R genes was found balanced on the three wheat sub-genomes. In contrast, at chromosome scale, 50 % of members of this gene family were localized on 6 of the 21 wheat chromosomes and ~22 % of them were localized on homeologous group 7. The results of this study provide a detailed analysis of the largest family of plant disease resistance genes in allohexaploid wheat. Some structural traits reported had not been previously identified and the genome-derived data were confronted with those stored in databases outlining the functional specialization of members of this family. The large reservoir of NBS-type genes presented and discussed will, firstly, form an important framework for marker-assisted improvement of resistance in wheat, and, secondly, open up new perspectives for a better understanding of the evolution dynamics of this gene family in grass species and in polyploid systems.
|
|
Scooped by
Kamoun Lab @ TSL
March 2, 2015 6:10 PM
|
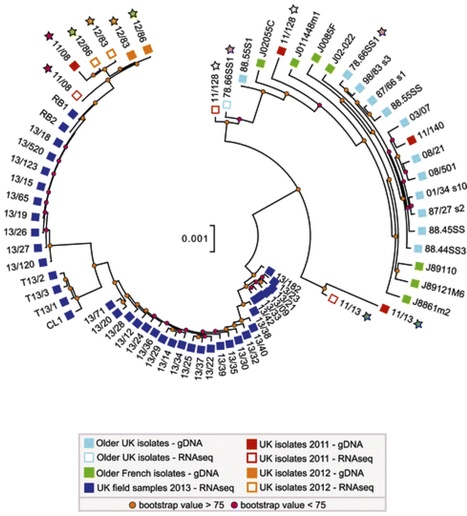
Field pathogenomics adds highly informative data to surveillance surveys by enabling rapid evaluation of pathogen variability, population structure and host genotype.
Yellow rust, caused by Puccinia striiformis f. sp. tritici (PST), is a major disease of wheat and, together with stem rust (Puccinia graminis) and leaf rust (Puccinia triticina), causes some of the most devastating epidemics on wheat worldwide [1]. Control of these rust pathogens relies predominantly on breeding and deployment of resistant varieties of wheat. To date, nearly 200 wheat-rust-resistance genes have been catalogued [2]; however, resistance has often proved to be ephemeral owing to changes in the pathogen population. In order to increase the durability of resistance, gene-deployment strategies need to consider extant and potential pathogen variability. Although these concepts are not new [3], their implementation was difficult until the advent of high-throughput sequencing (HTS) and genotyping technologies.
Next-generation sequencing technologies provide new opportunities to study pathogens and the hosts they infect. The increasing availability of crop and pathogen genomes [4] is providing new insights into pathogen biology, population structure and pathogenesis. This provides new opportunities for disease management. An important input into resistance breeding programs should be surveillance of the pathogen population. High-throughput pathogenomics offers the possibility for analyzing a large number of pathogen isolates and host varieties rapidly and at low cost.
In an article published in Genome Biology, Hubbard and colleagues [5] implemented a robust and rapid method to screen field isolates of PST and their host cultivars. In this particular version of pathogenomics, a selected set of 39 samples of infected wheat and triticale leaf tissue were collected directly from the field in 2013 and analyzed using RNAseq. In addition, the genomes of 21 archived PST isolates from the UK and France were also sequenced. Transcriptome analysis restricted the amount of sequence necessary to obtain diagnostic information for both host and pathogen; this not only accelerated genetic analysis of PST populations in situ but also allowed simultaneous assessment of the host genotype in the same sequencing runs. Another advantage of transcriptome analysis is that it detects genes being expressed and therefore the determinants of the interaction; thus, non-expressed genes present in the genome do not obscure genotype-phenotype correlations.
|
|
Scooped by
Kamoun Lab @ TSL
February 27, 2015 1:58 AM
|
Background Emerging and re-emerging pathogens imperil public health and global food security. Responding to these threats requires improved surveillance and diagnostic systems. Despite their potential, genomic tools have not been readily applied to emerging or re-emerging plant pathogens such as the wheat yellow (stripe) rust pathogen Puccinia striiformis f. sp. tritici (PST). This is due largely to the obligate parasitic nature of PST, as culturing PST isolates for DNA extraction remains slow and tedious. Results To counteract the limitations associated with culturing PST, we developed and applied a field pathogenomics approach by transcriptome sequencing infected wheat leaves collected from the field in 2013. This enabled us to rapidly gain insights into this emerging pathogen population. We found that the PST population across the United Kingdom, UK, underwent a major shift in recent years. Population genetic structure analyses revealed four distinct lineages that correlated to the phenotypic groups determined through traditional pathology-based virulence assays. Furthermore, the genetic diversity between members of a single population cluster for all 2013 PST field samples was much higher than that displayed by historical UK isolates, revealing a more-diverse population of PST. Conclusions Our field pathogenomics approach uncovered a dramatic shift in the PST population in the UK, likely due to a recent introduction of a diverse set of exotic PST lineages. The methodology described herein accelerates genetic analysis of pathogen populations and circumvents the difficulties associated with obligate plant pathogens. In principle, this strategy can be widely applied to a variety of plant pathogens.
|
|
Scooped by
Kamoun Lab @ TSL
February 25, 2015 9:17 AM
|
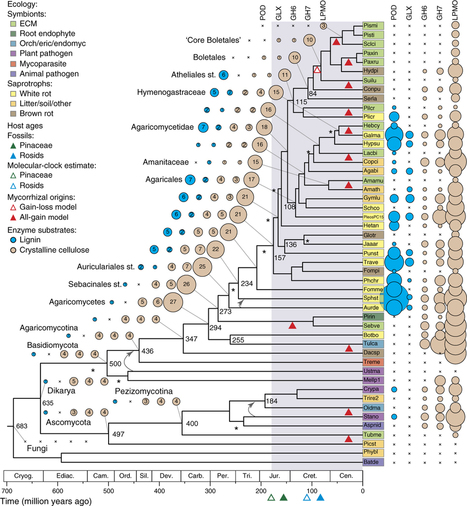
To elucidate the genetic bases of mycorrhizal lifestyle evolution, we sequenced new fungal genomes, including 13 ectomycorrhizal (ECM), orchid (ORM) and ericoid (ERM) species, and five saprotrophs, which we analyzed along with other fungal genomes. Ectomycorrhizal fungi have a reduced complement of genes encoding plant cell wall–degrading enzymes (PCWDEs), as compared to their ancestral wood decayers. Nevertheless, they have retained a unique array of PCWDEs, thus suggesting that they possess diverse abilities to decompose lignocellulose. Similar functional categories of nonorthologous genes are induced in symbiosis. Of induced genes, 7–38% are orphan genes, including genes that encode secreted effector-like proteins. Convergent evolution of the mycorrhizal habit in fungi occurred via the repeated evolution of a 'symbiosis toolkit', with reduced numbers of PCWDEs and lineage-specific suites of mycorrhiza-induced genes.
|
|
Scooped by
Kamoun Lab @ TSL
February 11, 2015 3:34 AM
|
Keynote presentation, 4th February 2015, León, México - part of the 2015 Genomics Research on Plant-Parasite Interactions to Increase Food Production UK-MX Workshop.
|
|
Scooped by
Kamoun Lab @ TSL
January 12, 2015 8:30 AM
|
Saprotrophic and parasitic microorganisms secrete proteins into the environment to breakdown macromolecules and obtain nutrients. The molecules secreted are collectively termed the ‘secretome’ and the composition and function of this set of proteins varies depending on the ecology, life cycle, and environment of an organism. Beyond the function of nutrient acquisition, parasitic lineages must also secrete molecules to counteract host defenses. Here we use a combination of de-novo genome and transcriptome sequencing and bioinformatic identification of signal peptides to identify the putative secreted proteome of two oomycetes, the facultative parasite Achlya hypogyna and free-living Thraustotheca clavata. By comparing the secretomes of these saprolegnialean oomycetes with that of 8 other oomycetes, we were able to characterize the evolution of this protein set across the oomycete clade. These species span the last common ancestor of the two major oomycete families allowing us to identify the ancestral secretome. This ancestral secretome consists of at least 84 gene families that encode putatively secreted proteins. Only 11 of these gene families are conserved across all 10 secretomes analysed and the two major branches in the oomycete radiation. Notably, we have identified expressed elicitin-like effector genes in the saprotrophic decomposer, T. clavata. Phylogenetic analyses show six novel HGTs to the oomycete secretome from bacterial and fungal donor lineages, four of which are specific to the Saprolegnialeans. Comparisons between free-living and pathogenic taxa highlight the functional changes of oomycete secretomes associated with shifts in lifestyle.
|
|
Scooped by
Kamoun Lab @ TSL
November 3, 2014 5:29 AM
|
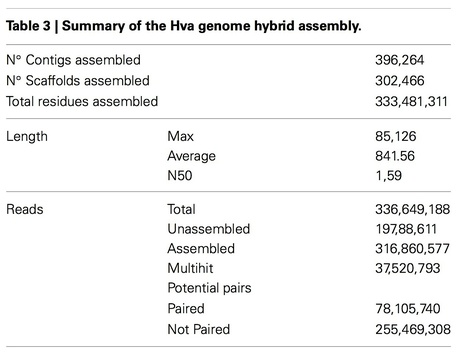
Coffee leaf rust caused by the fungus Hemileia vastatrix is the most damaging disease to coffee worldwide. The pathogen has recently appeared in multiple outbreaks in coffee producing countries resulting in significant yield losses and increases in costs related to its control. New races/isolates are constantly emerging as evidenced by the presence of the fungus in plants that were previously resistant. Genomic studies are opening new avenues for the study of the evolution of pathogens, the detailed description of plant-pathogen interactions and the development of molecular techniques for the identification of individual isolates. For this purpose we sequenced 8 different H. vastatrixisolates using NGS technologies and gathered partial genome assemblies due to the large repetitive content in the coffee rust hybrid genome; 74.4% of the assembled contigs harbor repetitive sequences. A hybrid assembly of 333 Mb was built based on the 8 isolates; this assembly was used for subsequent analyses. Analysis of the conserved gene space showed that the hybrid H. vastatrixgenome, though highly fragmented, had a satisfactory level of completion with 91.94% of core protein-coding orthologous genes present. RNA-Seq from urediniospores was used to guide the de novo annotation of the H. vastatrix gene complement. In total, 14,445 genes organized in 3921 families were uncovered; a considerable proportion of the predicted proteins (73.8%) were homologous to other Pucciniales species genomes. Several gene families related to the fungal lifestyle were identified, particularly 483 predicted secreted proteins that represent candidate effector genes and will provide interesting hints to decipher virulence in the coffee rust fungus. The genome sequence of Hva will serve as a template to understand the molecular mechanisms used by this fungus to attack the coffee plant, to study the diversity of this species and for the development of molecular markers to distinguish races/isolates.
|
|
Scooped by
Kamoun Lab @ TSL
September 23, 2014 4:16 AM
|
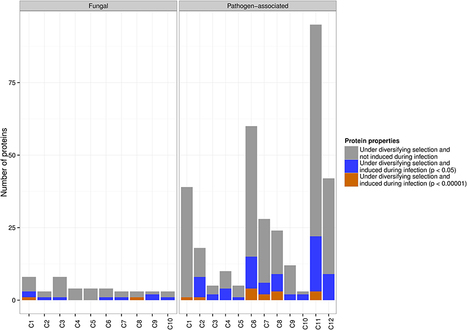
Plant pathogens cause severe losses to crop plants and threaten global food production. One striking example is the wheat stem rust fungus, Puccinia graminis f. sp. tritici, which can rapidly evolve new virulent pathotypes in response to resistant host lines. Like several other filamentous fungal and oomycete plant pathogens, its genome features expanded gene families that have been implicated in host-pathogen interactions, possibly encoding effector proteins that interact directly with target host defense proteins. Previous efforts to understand virulence largely relied on the prediction of secreted, small and cysteine-rich proteins as candidate effectors and thus delivered an overwhelming number of candidates. Here, we implement an alternative analysis strategy that uses the signal of adaptive evolution as a line of evidence for effector function, combined with comparative information and expression data. We demonstrate that in planta up-regulated genes that are rapidly evolving are found almost exclusively in pathogen-associated gene families, affirming the impact of host-pathogen co-evolution on genome structure and the adaptive diversification of specialized gene families. In particular, we predict 42 effector candidates that are conserved only across pathogens, induced during infection and rapidly evolving. One of our top candidates has recently been shown to induce genotype-specific hypersensitive cell death in wheat. This shows that comparative genomics incorporating the evolutionary signal of adaptation is powerful for predicting effector candidates for laboratory verification. Our system can be applied to a wide range of pathogens and will give insight into host-pathogen dynamics, ultimately leading to progress in strategies for disease control.
|
|
Scooped by
Kamoun Lab @ TSL
September 10, 2014 5:23 AM
|
Genome and Transcriptome of Mycorrhizal fungi - by S. Ghignone & R. Balestrini
|
|
|
Scooped by
Kamoun Lab @ TSL
August 15, 2015 7:06 AM
|
The oomycete Phytophthora infestans was the causal agent of the Irish Great Famine and is a recurring threat to global food security. The pathogen can reproduce both sexually and asexually and has a potential to adapt both abiotic and biotic environment. Although in many regions the A1 and A2 mating types coexist, the far majority of isolates belong to few clonal, asexual lineages. As other oomycetes, P. infestans is thought to be diploid during the vegetative phase of its life cycle, but it was observed that trisomy correlated with virulence and mating type locus and that polyploidy can occur in some isolates. It remains unknown about the frequency of polyploidy occurrence in nature and the relationship between ploidy level and sexuality. Here we discovered that the sexuality of P. infestans isolates correlates with ploidy by comparison of microsatellite fingerprinting, genome-wide polymorphism, DNA quantity, and chromosome numbers. The sexual progeny of P. infestans in nature are diploid, whereas the asexual lineages are mostly triploids, including successful clonal lineages US-1 and 13_A2. This study reveals polyploidization as an extra evolutionary risk to this notorious plant destroyer.
|
|
Scooped by
Kamoun Lab @ TSL
July 31, 2015 1:12 PM
|
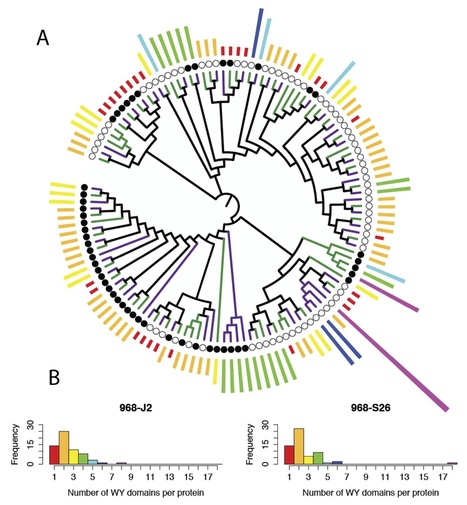
Peronospora tabacina is an obligate biotrophic oomycete that causes blue mold or downy mildew on tobacco (Nicotiana tabacum). It is an economically important disease occurring frequently in tobacco growing regions worldwide. We have sequenced and characterized the genomes of two P. tabacina isolates and mined them for pathogenicity related proteins and effector encoding genes. De novo assembly of the genomes using Illumina reads resulted in 4,016 (63.1 Mb, N50 = 79 kb) and 3,245 (55.3 Mb, N50 = 61 kb) scaffolds for isolates 968-J2 and 968-S26 respectively, with an estimated genome size of 68 Mb. The mitochondrial genome has a similar size (~43 kb) and structure to those of other oomycetes, plus several minor unique features. Repetitive elements, primarily retrotransposons, make up ~24% of the nuclear genome. Approximately 18 K protein coding gene models were predicted. Mining the secretome revealed ~120 candidate RxLR, ~6 CRN and ~61 WY-domain containing proteins. Candidate RxLR effectors were shown to be predominantly undergoing diversifying selection, with ~57% located in variable gene-sparse regions of the genome. Aligning the P. tabacina genome to Hyaloperonospora arabidopsidis and Phytophthora spp. revealed a high level of synteny. Blocks of synteny show gene inversions and instances of expansion in intergenic regions. Extensive rearrangements of the gene-rich genomic regions do not appear to have occurred during the evolution of these highly variable pathogens. These assemblies provide the basis for studies of virulence in this and other downy mildew pathogens.
|
|
Scooped by
Kamoun Lab @ TSL
July 3, 2015 11:37 AM
|
Fungi and oomycetes include deep and diverse lineages of eukaryotic plant pathogens. The last 10 years have seen the sequencing of the genomes of a multitude of species of these so-called filamentous plant pathogens. Already, fundamental concepts have emerged. Filamentous plant pathogen genomes tend to harbor large repertoires of genes encoding virulence effectors that modulate host plant processes. Effector genes are not randomly distributed across the genomes but tend to be associated with compartments enriched in repetitive sequences and transposable elements. These findings have led to the “two-speed genome” model in which filamentous pathogen genomes have a bipartite architecture with gene sparse, repeat rich compartments serving as a cradle for adaptive evolution. Here, we review this concept and discuss how plant pathogens are great model systems to study evolutionary adaptations at multiple time scales. We will also introduce the next phase of research on this topic.
|
|
Rescooped by
Kamoun Lab @ TSL
from Plants and Microbes
April 30, 2015 2:00 AM
|
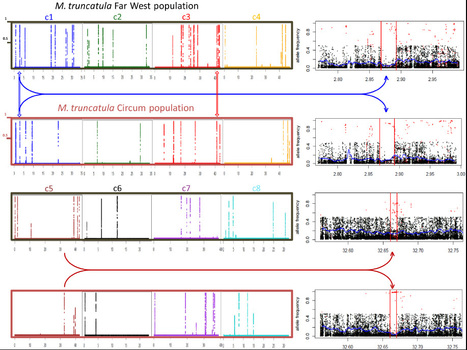
Medicago truncatula is a model legume species used to investigate plant-microorganism interactions, notably root symbioses. Massive population genomic and transcriptomic data now available for this species open the way for a comprehensive investigation of genomic variations associated with adaptation of M. truncatula to its environment. Here we performed a fine-scale genome scan of selective sweep signatures in Medicago truncatula using more than 15 million SNPs identified on 283 accessions from two populations (Circum and Far West), and exploited annotation and published transcriptomic data to identify biological processes associated with molecular adaptation. We identified 58 swept genomic regions with a 15 kb average length and comprising 3.3 gene models on average. The unimodal sweep state probability distribution in these regions enabled us to focus on the best single candidate gene per region. We detected two unambiguous species-wide selective sweeps, one of which appears to underlie morphological adaptation. Population genomic analyses of the remaining 56 sweep signatures indicate that sweeps identified in the Far West population are less population-specific and probably more ancient than those identified in the Circum population. Functional annotation revealed a predominance of immunity-related adaptations in the Circum population. Transcriptomic data from accessions of the Far West population allowed inference of four clusters of co-regulated genes putatively involved in the adaptive control of symbiotic carbon flow and nodule senescence, as well as in other root adaptations upon infection with soil microorganisms. We demonstrate that molecular adaptations in Medicago truncatula were primarily triggered by selective pressures from root-associated micro-organisms.
|
|
Rescooped by
Kamoun Lab @ TSL
from Plants and Microbes
March 11, 2015 7:32 AM
|
MPMI has played a leading role in disseminating new insights into plant-microbe interactions and promoting new approaches. Articles in this Focus Issue highlight the power of genomic studies in uncovering novel determinants of plant interactions with microbial symbionts (good), pathogens (bad), and complex microbial communities (unknown). Many articles also illustrate how genomics can support translational research by quickly advancing our knowledge of important microbes that have not been widely studied.
Click on Next Article or Table of Contents above to view the articles in this Focus Issue. (From the mobile site, go to the MPMI March 2015 issue.)
|
|
Scooped by
Kamoun Lab @ TSL
March 2, 2015 7:13 AM
|
How generalist parasites with wide host ranges can evolve is a central question in parasite evolution. Albugo candida is an obligate biotrophic parasite that consists of many physiological races that each specialize on distinct Brassicaceae host species. By analyzing genome sequence assemblies of five isolates, we show they represent three races that are genetically diverged by ~1%. Despite this divergence, their genomes are mosaic-like, with ~25% being introgressed from other races. Sequential infection experiments show that infection by adapted races enables subsequent infection of hosts by normally non-infecting races. This facilitates introgression and the exchange of effector repertoires, and may enable the evolution of novel races that can undergo clonal population expansion on new hosts. We discuss recent studies on hybridization in other eukaryotes such as yeast, Heliconius butterflies, Darwin's finches, sunflowers and cichlid fishes, and the implications of introgression for pathogen evolution in an agro-ecological environment.
|
|
Scooped by
Kamoun Lab @ TSL
February 25, 2015 9:22 AM
|
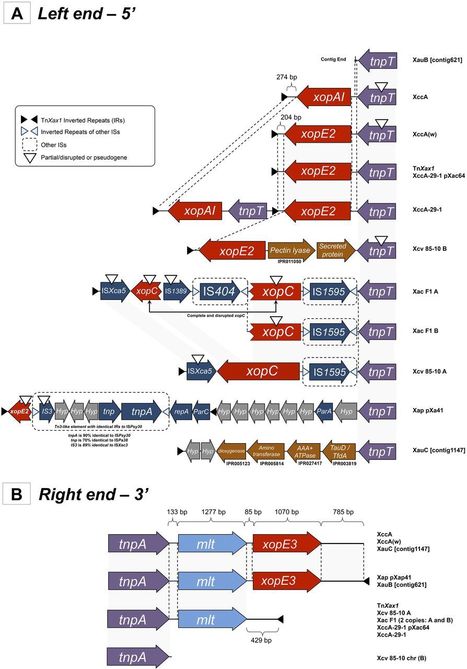
Members of the genus Xanthomonas are among the most important phytopathogens. A key feature of Xanthomonas pathogenesis is the translocation of type III secretion system (T3SS) effector proteins (T3SEs) into the plant target cells via a T3SS. Several T3SEs and a murein lytic transglycosylase gene (mlt, required for citrus canker symptoms) are found associated with three transposition-related genes in Xanthomonas citri plasmid pXAC64. These are flanked by short inverted repeats (IRs). The region was identified as a transposon, TnXax1, with typical Tn3 family features, including a transposase and two recombination genes. Two 14-bp palindromic sequences within a 193-bp potential resolution site occur between the recombination genes. Additional derivatives carrying different T3SEs and other passenger genes occur in different Xanthomonas species. The T3SEs include transcription activator-like effectors (TALEs). Certain TALEs are flanked by the same IRs as found in TnXax1 to form mobile insertion cassettes (MICs), suggesting that they may be transmitted horizontally. A significant number of MICs carrying other passenger genes (including a number of TALE genes) were also identified, flanked by the same TnXax1 IRs and delimited by 5-bp target site duplications. We conclude that a large fraction of T3SEs, including individual TALEs and potential pathogenicity determinants, have spread by transposition and that TnXax1, which exhibits all of the essential characteristics of a functional transposon, may be involved in driving MIC transposition. We also propose that TALE genes may diversify by fork slippage during the replicative Tn3 family transposition. These mechanisms may play a crucial role in the emergence of Xanthomonas pathogenicity.
|
|
Scooped by
Kamoun Lab @ TSL
February 11, 2015 7:59 AM
|
Background Many plant-pathogenic fungi have a tendency towards genome size expansion, mostly driven by increasing content of transposable elements (TEs). Through comparative and evolutionary genomics, five members of the Leptosphaeria maculans-Leptosphaeria biglobosa species complex (class Dothideomycetes, order Pleosporales), having different host ranges and pathogenic abilities towards cruciferous plants, were studied to infer the role of TEs on genome shaping, speciation, and on the rise of better adapted pathogens.Results L. maculans ‘brassicae’, the most damaging species on oilseed rape, is the only member of the species complex to have a TE-invaded genome (32.5%) compared to the other members genomes (<4%). These TEs had an impact at the structural level by creating large TE-rich regions and are suspected to have been instrumental in chromosomal rearrangements possibly leading to speciation. TEs, associated with species-specific genes involved in disease process, also possibly had an incidence on evolution of pathogenicity by promoting translocations of effector genes to highly dynamic regions and thus tuning the regulation of effector gene expression in planta.Conclusions Invasion of L. maculans ‘brassicae’ genome by TEs followed by bursts of TE activity allowed this species to evolve and to better adapt to its host, making this genome species a peculiarity within its own species complex as well as in the Pleosporales lineage.
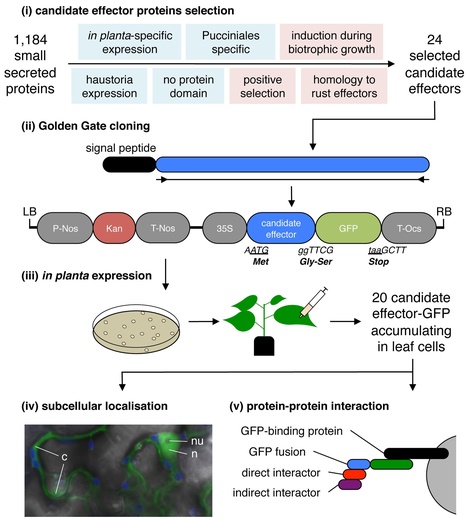
Rust fungi are devastating crop pathogens that deliver effector proteins into infected tissues to modulate plant functions and promote parasitic growth. The genome of the poplar leaf rust fungus Melampsora larici-populina revealed a large catalogue of secreted proteins, some of which have been considered candidate effectors. Unravelling how these proteins function in host cells is key to understanding pathogenicity mechanisms and developing resistant plants. In this study, we used an effectoromics pipeline to select, clone, and express 20 candidate effectors in Nicotiana benthamiana leaf cells to determine their subcellular localisation and identify the plant proteins they interact with. Confocal microscopy revealed that six candidate effectors target the nucleus, nucleoli, chloroplasts, mitochondria and discrete cellular bodies. We also used coimmunoprecipitation and mass spectrometry to identify 606 N. benthamiana proteins that associate with the candidate effectors. Five candidate effectors specifically associated with a small set of plant proteins that may represent biologically relevant interactors. We confirmed the interaction between the candidate effector MLP124017 and the TOPLESS-Related Protein 4 from poplar by in planta coimmunoprecipitation. Altogether, our data enable us to validate effector proteins from M. larici-populina and reveal that these proteins may target multiple compartments and processes in plant cells. It also shows that N. benthamiana can be a powerful heterologous system to study effectors of obligate biotrophic pathogens.
Via The Sainsbury Lab
An individual of the mushroom-forming fungus Armillaria bulbosa is among the largest and oldest of all living organisms: More than 1500 years old, it covers more than 15 ha and weighs more than 10,000 kg (1). Some trees can also reach ages of thousands of years (2). How can such long-lived organisms keep the number of deleterious mutations during somatic growth in check? In a recent paper in Mycologia, Anderson and Catona (3) report extremely low genetic variation, and by inference a very low mutation rate, in a long-lived individual of another fungus, Armillaria gallica (see the photo). This genomic stability is puzzling and unexpected, because the sequenced samples come from locations that are more than 100 m apart and presumably separated by many rounds of cell division.
Via Francis Martin
|
|
Scooped by
Kamoun Lab @ TSL
October 28, 2014 6:59 AM
|
Advances in genome-based studies on plant-associated microorganisms have transformed our understanding of many plant pathogens and are beginning to greatly widen our knowledge of plant interactions with mutualistic and commensal microorganisms. Pathogenomics has revealed how pathogenic microorganisms adapt to particular hosts, subvert innate immune responses and change host range, as well as how new pathogen species emerge. Similarly, culture-independent community profiling methods, coupled with metagenomic and metatranscriptomic studies, have provided the first insights into the emerging field of research on plant-associated microbial communities. Together, these approaches have the potential to bridge the gap between plant microbial ecology and plant pathology, which have traditionally been two distinct research fields.
|
|
Scooped by
Kamoun Lab @ TSL
September 11, 2014 10:22 AM
|
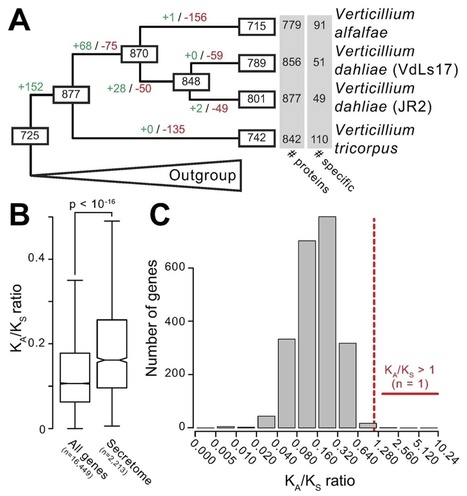
Vascular wilts caused by Verticillium spp. are destructive plant diseases, affecting hundreds of hosts. Only few Verticillium spp. are causal agents of vascular wilt diseases, of which V. dahliae is the most notorious pathogen, and several V. dahliae genomes are available. In contrast, V. tricorpus is mainly known as saprophyte and causal agent of opportunistic infections. Based on a hybrid approach that combines second and third generation sequencing, a near-gapless V. tricorpus genome assembly was obtained. With comparative genomics, we aimed to identify genomic features in V. dahliae that confer the ability to cause vascular wilt disease. Unexpectedly, both species encode similar effector repertoires and share a genomic structure with genes encoding secreted proteins clustered in genomic islands. Intriguingly, V. tricorpus contains significantly less repetitive elements and an extended spectrum of secreted carbohydrate-active enzymes when compared with V. dahliae. In conclusion, we highlight the technical advances of a hybrid sequencing and assembly approach and reveal that the saprophyte V. tricorpus shares many hallmark features with V. dahliae.
|