 Your new post is loading...
Your new post is loading...

|
Scooped by
Jean-Michel Ané
Today, 9:45 AM
|
Background
The trans-kingdom movement of microRNAs (miRNAs) is a key regulatory mechanism in plant biotic interactions, influencing and modulating both pathogenic virulence and symbiotic relationships. This study employed bioinformatic analysis to predict the role of rice (Oryza sativa L.) miRNAs in regulating gene expression during interactions with the pathogenic fungi Rhizoctonia solani (R. solani) and Magnaporthe oryzae (M. oryzae), as well as the symbiotic fungus Rhizophagus irregularis (R. irregularis).
Results
Previously reported and up-regulated rice miRNAs were identified from the literature during inoculation, and their target genes were predicted in the fungal transcriptomes. The KEGG pathway and GO enrichment analyses revealed that rice miRNAs could target genes crucial for pathogen metabolism (e.g., carbohydrate, amino acid, lipid, and xenobiotic), genetic information processing, and essential cellular processes (e.g., signal transduction, transport, catabolism, and cell growth and death), potentially impairing fungal virulence and survival. The osa-miR171 family is predicted to regulate R. irregularis genes involved in protein serine/threonine kinase activity, antiporter activity, cell division, TAP complex binding, and O-acetylhomoserine sulfhydrylase activity during symbiosis. Additionally, the osa-miR171 family targets key rice genes such as phosphate transporter, NSP2, and SCR, involved in nutrient transport, common symbiosis signaling, and root development, respectively, which are likely important for forming and regulating a symbiotic relationship. The qRT‒PCR results confirmed the up-regulation of osa-miR171h (symbiosis-related) and osa-miR167d-5p (pathogen-responsive) in rice roots inoculated with R. irregularis.
Conclusions
These findings highlight the significant role of trans-kingdom RNA regulation in plant–microbe interactions, contributing to understanding the molecular regulation, distinctions, and similarities between pathogenic and symbiotic relationships in plants. This initial study performs the first comparative trans-kingdom sRNA analysis in rice, successfully predicting and distinguishing the molecular mechanisms that drive virulence versus mutualism. This work also lays the groundwork for developing novel strategies to enhance plant disease resistance, foster beneficial symbiotic interactions, and ultimately boost agricultural yields.
|
|
Scooped by
Jean-Michel Ané
December 5, 11:14 AM
|
In northern biomes, growth is nitrogen (N) limited, but bryophytes are abundant. These bryophytes often host N2-fixing microorganisms (diazotrophs) that play a crucial role in the N cycle of these ecosystems. Despite their importance, how the bryophyte-associated N2-fixation varies across species and seasons (summer, autumn, winter, and spring) remains poorly understood. We measured N2-fixation rates for 10 bryophyte species in situ throughout the entire year in the Arctic with additional incubations to verify the method. We measured positive N2-fixation during most of the year, except for the coldest period (February). The species growing in the wettest conditions (Sphagnum spp.) had the highest N2-fixation rates in summer, while bryophytes in drier conditions peaked in N2-fixation rates in spring and autumn. The seasonal variation in N2-fixation activity was pronounced, but similar patterns were found among different species. This study reveals that bryophyte-associated N2-fixation in northern ecosystems is larger than previously assumed, as it occurs over a more extended part of the year than previously inferred. Furthermore, the importance of bryophyte-associated diazotrophs cannot be quantified without considering both the diversity of bryophytes and their variation in N2-fixing seasonal activity patterns. Both future changes in climatic conditions and biodiversity of bryophytes can thus have large implications for the N cycle in arctic regions.
|
|
Scooped by
Jean-Michel Ané
December 5, 11:02 AM
|
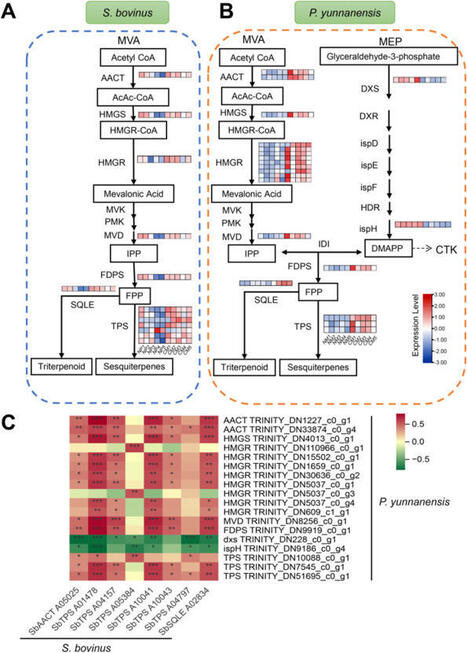
Suillus bovinus is an ecologically significant wild ectomycorrhizal fungus that forms symbiotic associations with pine trees. Terpenoids are known to play a key role in interactions between fungi and plants. However, the molecular mechanisms underlying the effects of terpenoids on the formation of symbiotic associations with S. bovinus remain poorly understood. Here, we investigated the effect of S. bovinus terpenoids on root development and mycorrhizal formation in Pinus yunnanensis using physiological, transcriptomic, and phytohormone assays. Our findings revealed that terpenoids from S. bovinus were associated with alterations in the synthesis and signaling of cytokinin in P. yunnanensis, evidenced by changes in the expression of specific genes in the mevalonate synthesis pathway and signaling transduction, such as CKXs, AHP, and A-ARRs. Similarly, these terpenoids correlated with changes in auxin synthesis and signaling, indicated by modulated tryptophan levels and the expression of specific enzyme genes, such as GH3, AUX1, AUX/IAA, and SAUR. This suggests that terpenoids from S. bovinus may contribute to lateral root development and mycorrhizal formation in P. yunnanensis, potentially by influencing the relative levels of cytokinin and auxin. These results shed light on the potential involvement of terpenoids produced by S. bovinus in establishing symbiotic interactions between ectomycorrhizal fungi and their host plants, warranting further direct functional validation.
|
|
Scooped by
Jean-Michel Ané
December 4, 5:02 PM
|
it is thought that the aerial brace roots can associate with nitrogen-fixing bacteria
|
|
Scooped by
Jean-Michel Ané
December 2, 7:12 AM
|
Heterosis, or hybrid vigor, describes the superior performance of F1 hybrids compared to parental inbreds. While soil microbiomes are proposed to influence heterosis, it remains unclear how heterotic plants shape their microbiomes and how interactions relate to stress responses. Here, we investigate the role of rhizosheath formation—the soil tightly adhering to roots—in maize heterosis under nitrogen deprivation. Across sterilization, inoculation, and transplantation experiments, hybrids develop larger rhizosheaths than inbreds, and rhizosheath size associates with biomass heterosis. Rhizosheath-enriched genus Massilia correlates with lateral root density, rhizosheath size, and growth. Untargeted metabolomics and flavone-deficient mutants reveal links between Massilia and flavonoid pathways, while growth promotion by Massilia can also occur independently of host flavones. Metagenomic analysis shows that larger rhizosheaths recruit microbial functions related to nutrient cycling and stress adaptation. These findings identify rhizosheath formation as an integrative trait associated with heterosis and a promising target for breeding resilient crops.
|
|
Scooped by
Jean-Michel Ané
December 2, 7:02 AM
|
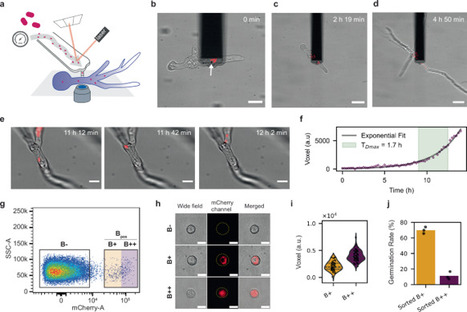
Endosymbioses represent dynamic relationships between organisms that may involve antagonistic phases during their emergence. Here, we induced cell-in-cell interactions between the free-living bacterium Ralstonia pickettii and an endosymbiont-free strain of the fungus Rhizopus microsporus using fluidic force microscopy to investigate the early phase of endosymbiosis formation. Following the implantation of bacteria into the cytosol, the rapid proliferation of R. pickettii compromised host fitness, as evidenced by reduced fungal viability, and triggered immune responses characterized by upregulated expression of stress-related defense genes. Vertical transmission of bacteria across fungal generations enabled repeated rounds of selective passaging, ultimately resulting in transcriptional relaxation of the fungal defense response. High-throughput-imaging revealed that the propagated system accommodated higher bacterial loads within viable spores, with a corresponding reduction in fungal growth. The observed physiological changes and comparative fungal transcriptomic profiles indicated adaptive resilience and a shift from antagonism to commensalism. This transition was characterized by attenuated expression of genes involved in cell wall remodeling and reactive oxygen metabolism. Our experimental system provides insights into the early processes of endosymbiosis, supporting the hypothesis that facultative intracellular pathogens can serve as intermediates toward stable endosymbiotic relationships.
|
|
Scooped by
Jean-Michel Ané
November 30, 9:45 PM
|
Interactions between arbuscular mycorrhizal (AM) fungi and ammonia-oxidizing (AO) microorganisms, two important microbial guilds contributing to soil-plant mineral nutrient cycling, are complex, given the high variability of soil biological, physical, and chemical properties. In addition, AO microorganisms are generally slow growing and require ample time to establish. Their communities are thus difficult to reconstruct under laboratory conditions, for example after soil sterilization. Therefore, in this study, we investigated quantitative and compositional responses of indigenous microorganisms occurring in 50 different field soils (collected from grasslands and arable fields) to actively growing mycelium of the AM fungus Rhizophagus irregularis. To this end, we quantified the abundance of various microbial guilds including AO bacteria (AOB), AO archaea (AOA), and comammox Nitrospira in pot-incubated soils exposed or not to actively growing AM fungus. Across the variety of soils, we observed systematic suppression by the AM fungus of different microbial groups including bacteria, protists, and fungi. The strongest suppression was noted for AOB and comammox Nitrospira, whereas the abundance and community structure of AOA remained unaffected by the AM fungal activity. Mycorrhizal suppression of AOB abundance was accompanied by changes in AOB community structure and correlated with soil pH. Contrary to the expected competition between AM fungus and AO microorganisms for available ammonium (NH4+) in the soil solution, the presence of the actively growing AM fungus significantly increased soil NH4+ levels as compared to the non-mycorrhizal control, at least upon the final destructive harvest. Thus, the interaction between the AM fungi and AO microorganisms likely goes beyond the simple competition for the free ammonium ions and might involve microorganisms active in other pathways of soil nitrogen cycle (e.g., mineralization) or temporarily different trajectories of nutrient use in mycorrhizal vs. non-mycorrhizal systems. Alternatively, elusive biological nitrification inhibitors may have contributed to the observed effect, produced by the AM fungus or its host plant, and subsequently transported to the root-free soil via the AM fungal hyphae.
|
|
Scooped by
Jean-Michel Ané
November 30, 9:36 PM
|
Alpine plants in nitrogen-deficient environments can acquire nitrogen by associating with endophytic nitrogen-fixing microorganisms that inhabit their roots and leaves to form symbiotic relationships. However, research is limited on nitrogen-fixing bacterial communities in the roots and leaves of alpine grassland plants, especially regarding the differences between various plant parts. In this study, we compared the root and leaf bacterial communities of four alpine plant families (Asteraceae, Leguminosae, Poaceae, and Rosaceae) in the alpine meadow ecosystem of Naqu, Tibet, using culture-based methods, 16S rRNA, and nifH gene pyrosequencing. The results showed greater bacterial diversity in the root compared to the leaf, and Fabaceae plants harbored a higher abundance of nitrogen-fixing bacteria. Interestingly, the roots and leaves of non-Fabaceae plants (Kobresia, Festuca ovina, and Leontopodium) also harbored abundant nitrogen-fixing communities such as Microbacterium, Curtobacterium, and Rhodococcus. Compared with subtropical environments, Cyanobacteria are important symbiotic nitrogen-fixing bacteria in plants of alpine ecosystems. These findings indicate that plant species and plant parts strongly influence the selection of bacterial populations. Understanding these microbial ecological functions in alpine grasslands provides scientific insights for optimizing agricultural practices and ecosystem management.
|
|
Scooped by
Jean-Michel Ané
November 30, 8:53 PM
|
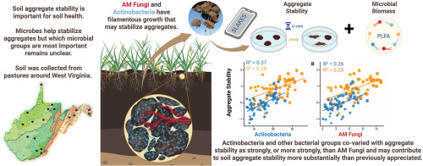
•Macroaggregate stability covaried most strongly with soil organic matter
•Actinobacteria moderately outperformed AMF as a predictor of soil macroaggregate stability
•Bacterial PLFAs more strongly covaried with stability than AMF in Appalachian pastures
•Penalized regression ranked bacterial groups as better predictors of stability over AMF
•Findings raise questions about long-held assumptions regarding AMF as primary soil stabilizers
|
|
Scooped by
Jean-Michel Ané
November 25, 2:00 PM
|
The NIN-like protein 2 (NLP2) transcription factor is highly expressed in the infected cells of the N2 fixation zone of mature nodules in Medicago truncatula. In these cells, NLP2 directly activates the expression of leghemoglobin genes and is required for the full expression of nitrite reductase (NiR). Histological examination of nodules formed under different nitrate regimes revealed that the infected cells of nlp2 display abnormal vacuole morphology under high nitrate (5 mM KNO3). This phenotype is associated with starch accumulation, higher expression of starch biosynthesis genes, and increased levels of nitrite and nitric oxide. A transcriptomic comparison of nlp2 and wild-type nodules across nitrate concentrations revealed that under high nitrate, nlp2 shows lower expression of genes important for hypoxia adaptation, such as alcohol dehydrogenase and pyruvate decarboxylase (PDC), and increased expression of S-nitrosoglutathione reductase (GSNOR), which encodes a key enzyme for nitric oxide homeostasis. These changes in gene expression were associated with lower nodule ATP levels and a higher NAD+/NADH ratio, suggesting that energy metabolism is specifically compromised in nlp2 nodules under high nitrate. Transgenic expression of NiR in the nodules of nlp2-1 mutants grown under high nitrate rescued the vacuole phenotype and restored the expression of GSNOR and PDC to normal levels. Overall, our data indicate a role for NLP2 in the regulation of NiR in N2-fixing cells, suggesting a role for nitrate reduction in nodule energy homeostasis.
|
|
Scooped by
Jean-Michel Ané
November 24, 6:07 PM
|
The root microbiota plays a vital role in plant growth and health. Recently, Zhang and colleagues demonstrated that the rhizobacterial strain Exiguobacterium R2567 produces cyclo(Leu-Pro), a cyclic dipeptide that regulates tillering by activating the rice strigolactone (SL) signaling pathway through binding to the SL receptor OsD14. This discovery provides an innovative strategy for optimizing crop architecture by harnessing the root microbiota for sustainable agriculture. It holds promise for achieving precision and environmentally friendly production through the application of synthetic microbial communities (SynComs) or functional metabolites. However, practical implementation faces challenges, including the field stability of cyclo(Leu-Pro), competition from native microbial communities, and potential long-term ecological risks, which require further study to realize its full potential.
|
|
Scooped by
Jean-Michel Ané
November 24, 2:06 PM
|
Iron is a critical micronutrient for both plants and humans, yet its declining availability across agricultural systems threatens global food security and health. Biofortification of food crops has emerged as a promising strategy to combat iron deficiency and anemia, leveraging both crop breeding and microbiome-based approaches to enhance iron mobilization and uptake. Advances in plant and bacterial synthetic biology could enable the precise programming of iron homeostasis and acquisition mechanisms, offering tailored solutions across diverse species and environments. Here, we outline key biomolecules, genes, and biosynthetic and transport pathways that represent underexplored synthetic biology targets for improving crop iron acquisition. We highlight opportunities to tune expression strength, tissue specificity, and cross-host pathway transfer to enhance chelation- and reduction-mediated solubilization of soil iron and augment plant uptake. Finally, we emphasize the broader importance of developing plant–microbe–metal actuators as modular components in genetic circuit design and discuss how their deployment across diverse plant and microbial chassis could accelerate agricultural biofortification and improve global nutrition.
|
|
Scooped by
Jean-Michel Ané
November 24, 1:07 PM
|
The root microbiome is important for plant development. The impact of the root microbiome is the sum of multiple complex interactions among microorganisms, the plant and the environment. This complexity can be reduced by designing synthetic bacterial communities (SynComs) consisting of bacteria isolated from the roots, making it possible to study these interactions. However, the translational power from SynCom experiments to explain field observations is still very low, which demonstrates the need for development of SynComs that colonize plants comparable to what is observed in the field. Hence, we developed a SynCom consisting of 13 different strains from 13 genera with varying phenotypes originating from the roots of winter wheat (Triticum aestivum cv. Sheriff). The SynCom was inoculated into gamma-irradiated soil prior to sowing and community assembly was determined over 4 weeks using 16S rRNA amplicon sequencing. The SynCom supported growth of winter wheat over a 4-week period and developed in a comparable manner to a more diverse natural community (NatCom) obtained from a soil solution. Furthermore, the temporal dynamics of the majority of the SynCom strains mimicked the development in relative abundance of their respective genera in field grown winter wheat of similar cultivar. However, this could not be translated to a different cultivar (Herup). Our results demonstrate how SynComs inoculated into gamma-irradiated soil can provide a framework for bridging the gap between greenhouse and lab experiments and field observations. At the same time it highlights the plant-genotype specific impact on community assembly.
|
|
|
Scooped by
Jean-Michel Ané
Today, 9:41 AM
|
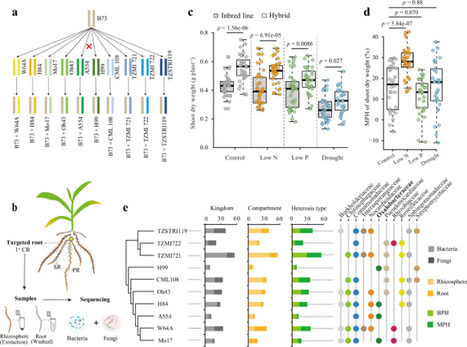
Improving crop nitrogen (N) uptake is essential for a more sustainable agriculture. Deploying resource-efficient root phenotypes, beneficial soil microbiomes and their interplay is a promising approach. To test the hypothesis that adaptive root phenotypes under N limitation associate to specific microbial taxa, we characterized 11 architectural and 13 anatomical root phenotypes, and associated rhizosphere prokaryotic and fungal communities across 16 field-grown maize (Zea mays L.) inbred lines under optimal and low N availability. While maize genotypes were not significant to the rhizosphere microbial diversity, the number of crown roots significantly affected fungal β-diversity under N limitation. Moreover, the relative abundance of 98 rhizosphere microbial taxa significantly correlated with individual root architectural or anatomical phenotypes in a N- and plant yield-specific way. Interestingly, a greater number of correlations was found under optimal than under low N availability. Our results suggest the importance of investigating the root phenotypes as predictors of rhizosphere microbial communities in maize inbred lines. Moreover, maize root architecture and anatomy may associate with microbes more frequently under optimal than under suboptimal N conditions. The relative contribution of root phenotypes and specific microbial taxa to plant performance under N limitation deserves more attention in future research.
|
|
Scooped by
Jean-Michel Ané
December 5, 11:04 AM
|
The compositions of conserved gene families often vary widely between species, complicating predictions and experimental tests of shared versus distinct functions, especially in families shaped by extensive duplication, redundancy, and paralog diversification. The plant CLV3/EMBRYO-SURROUNDING REGION (CLE) small signaling peptide family exemplifies these challenges. Although genetic studies in model systems have identified shared roles for a few CLE genes and species-specific redundancies, an evolutionary analysis of the entire family over deep time could empower predictive and experimental dissections of functions obscured by redundancy. We developed a scanning pipeline that de novo annotated CLE genes from 2,000 genomes representing 1,000 species, uncovering thousands of previously undetected family members and producing a comprehensive view of the family's evolution and sequence diversification over 140 million years. Computational modeling of coding and cis-regulatory regions predicted lineage-specific asymmetries in paralog redundancy, stemming from ancestral amino acids in the functional core of the dodecapeptide and partial conservation of promoter elements. We tested these predictions using two genome-editing strategies in Solanaceae. Base-editing of deeply conserved residues in the CLV3 dodecapeptide and its paralogs across three species confirmed their critical roles in repressing stem cell proliferation, and multiplex CRISPR knockouts of the 52 tomato CLE genes resolved simple and complex redundancies, revealing previously uncharacterized regulators of shoot architecture and plant size. These findings show how both peptide and cis-regulatory erosion shape CLE redundancy and provide a framework for detecting and translating deep evolutionary signals into testable genetic hypotheses across compositionally complex gene families.
|
|
Scooped by
Jean-Michel Ané
December 5, 10:57 AM
|
This research identified and validated a gene encoding the ATP synthase β subunit (AhatpB), which positively regulates both nodule number and the abundance of the microsymbiont Bradyrhizobium ASV1. Varieties carrying superior AhatpB haplotype exhibited higher pod yields. Compared with the nodules of varieties carrying the superior AhatpB haplotypes, the transcription and metabolism of oxidative phosphorylation were inhibited during nodule development in varieties carrying normal AhatpB haplotypes, leading to the accumulation of adenosine-5′-diphosphate, dihydrogen phosphate, and succinic acid, which were insufficient to maintain Bradyrhizobium growth and promote nodule development. Additionally, four endophytes (ASV7696, ASV4913, ASV1693, and ASV495) were found to be significantly positively correlated with nodule number, and most of the genes regulating their abundance encoded senescence-associated proteins (Arahy.4RGJ1T, Arahy.AA4TUV, Arahy.74ZQZH, Arahy.8PDB05). Furthermore, candidate genes (Arahy.FQH7NX, Arahy.2C7VNA, Arahy.T63ME6, Arahy.X7L1IJ, Arahy.JBBQ4L, and Arahy.IT8ZEZ) that regulate the abundance of endophytes significantly negatively correlated with nodule number were detected.
|
|
Scooped by
Jean-Michel Ané
December 2, 7:14 AM
|
Arbuscular mycorrhizal fungi (AMF) form symbiotic relationships with roots of most terrestrial plants, playing a crucial role in regulating soil organic carbon (SOC) dynamics. While precipitation increase (Pi) is a major facet of climate change, its impacts on root- and AMF-mediated SOC formation and stability remain largely unexplored. Here, we combined a meta-analysis across global grasslands with a multiyear precipitation manipulation experiment in a semiarid grassland on the Loess Plateau to disentangle the relative effects of roots and their associated AMF on microbial communities and SOC as influenced by Pi. We show that Pi induced tradeoffs between roots and AMF, and promoted SOC formation and stability via the mycelium- rather than the root-pathway, leading to an increase of 136% (±40) and 297% (±90) in mycelium-derived C and mineral-associated organic C (MAOC), respectively. Pi altered plant community composition, favoring subshrubs and forbs over grasses. Also, Pi reduced specific root length, but increased root diameter, tissue density, and root colonization and extraradical biomass of AMF. Furthermore, Pi-induced change in AMF shifted the soil bacterial community by favoring K-strategists, increasing bacterial necromass C and promoting MAOC accumulation. Our findings provide direct evidence that Pi enhances AMF-driven SOC sequestration by expanding the mycorrhizosphere and promoting microbiota with high C use efficiency, highlighting a key mechanism by which mycorrhizal fungi mediate SOC formation and stability under shifting precipitation regimes.
|
|
Scooped by
Jean-Michel Ané
December 2, 7:07 AM
|
The plant rhizosphere, a region interconnecting roots, soil, and microorganisms, is critical for plant resource acquisition, community structure, and the functional stability of ecosystems. Most studies focus primarily on root traits, while overlooking the covariation within the rhizosphere root–soil–microbe continuum and its ecological implications under environmental change. Here, we highlight the necessity of integrating rhizosphere function into a broader theoretical framework encompassing belowground traits, such as the core functional modules of roots, rhizosphere microorganisms (including mycorrhizal fungi), and soil. We further identify critical knowledge gaps and future directions for research on rhizosphere function traits. This framework expands current perspectives on plant belowground functional traits, plant adaptation, and ecosystem stability under changing environments.
|
|
Scooped by
Jean-Michel Ané
November 30, 9:46 PM
|
In recent years, interest in the role of nutrient cycling in sustainable agriculture has significantly increased. The potential of arbuscular mycorrhizal (AM) fungi (AMFs) in nutrient cycling and plant growth improvement has long been recognized. However, there have been only a few studies on the identification and exploration of AM symbiosis-related plant genes for sustainable agriculture. We have developed a new constructive model for using host plant-derived AM symbiosis-related genes to improve breeding and AMF utilization for sustainable agriculture, particularly in the context of climate change. This model include: 1) the discovery of AM symbiosis-related genes in crop wild-relatives for molecular breeding and 2) the screening and propagation of AMFs that can help improve water-use efficiency and nutrient-use efficiency by crops, thereby reducing chemical fertilizer use in agricultural production. The first approach uniquely facilitates the identification of host plant-derived AM symbiosis-related genes, such as CHITIN ELICITOR RECEPTOR KINASE 1 (OsCERK1) from Dongxiang (DY) wild rice (Oryza rufipogon) (OsCERK1DY), MILDEW RESISTANCE LOCUS 1 (MLO1) from wild barley (Hordeum spontaneum), and WRKY60 from wild soybean (Glycine soja), for breeding purposes. The second one involves identifying soil-borne AMF species, such as Rhizophagus intraradices and Glomus mosseae for practical applications in the field. This suggestive model presents an emerging biotechnological potential for engineering climate-resilient crops.
|
|
Scooped by
Jean-Michel Ané
November 30, 9:38 PM
|
Crop rotation is widely practiced to improve agricultural sustainability, yet its impact on microbial diversity remains unclear. We conduct a global meta-analysis of 2406 paired observations to examine the effects of crop rotation on microbial diversity based on high-throughput sequencing data. We show that crop rotation significantly increases bacterial Shannon diversity and species richness, but has no effect on bacterial beta diversity. In contrast, crop rotation significantly increases fungal beta diversity without affecting fungal Shannon diversity and species richness. Changes in microbial communities are linked to soil pH, and available nitrogen and phosphorus. Notably, legume vs. non-legume, arbuscular mycorrhiza vs. non- arbuscular mycorrhiza, C3 vs. C4, and annual vs. perennial crop transitions, as well as climate and soil factors, affect the response ratios of microbial metics to crop rotation. Furthermore, the response ratios of bacterial Shannon diversity, bacterial species richness, and fungal species richness are positively related to the response ratio of crop yield. Our study reveals positive but differential effects of crop rotation on bacterial and fungal diversities, which are linked to improved crop productivity. Our findings thus have implications for using crop rotation to conserve soil microbial biodiversity, which is related to soil health and function, and enhance global food security.
|
|
Scooped by
Jean-Michel Ané
November 30, 8:55 PM
|
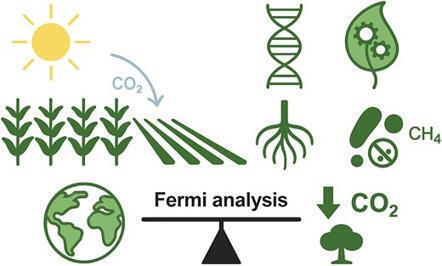
Plant agriculture contributes substantially to global greenhouse gas emissions, yet it also offers powerful opportunities for climate change mitigation. Here, we focus on how to identify and prioritize synthetic biology strategies to reduce emissions and sequester carbon through plant-based interventions. Effective solutions must process large volumes of carbon, be scalable, yield a positive life-cycle balance, and be economically viable, technically feasible, and deployable in field conditions without undue damage to what remains of nature on Earth. Using Fermi estimation, we quantify the per-hectare, annual, and 100-year CO2-equivalent (CO2e) drawdown potential of emerging synthetic biology strategies—including improved CO2 fixation, reduced yield losses, root-deposited biopolymers, engineered nitrogen fixation, and methane reduction—and benchmark them against nonengineered approaches such as biochar, forestation, and fast-growing biomass crops. We used a 100-year horizon to allow for both development and implementation of high-risk but high-impact synthetic biology strategies. We integrate factors such as per-hectare effectiveness, year-on-year sequestration, deployment area, and storage durability. We demonstrate that while per-hectare impacts vary by orders of magnitude (<1 to >30 t CO2e/ha/year), deployment scale is the dominant factor determining total impact. Targeted synthetic biology strategies implemented across existing agricultural systems could deliver ∼120 Gt CO2e drawdown over a century and contribute to an additional ∼140 Gt CO2e drawdown. Decreasing synthetic nitrogen fertilizer use and biochar implementation have the biggest CO2e impact potential. Early-stage quantitative evaluation is critical to guide R&D toward climate-relevant solutions and deliver a prioritized portfolio of near- and long-term strategies. A transdisciplinary approach—linking synthetic biology, agronomy, engineering, and social systems—is essential to realize impact. This work offers a framework for evaluating plant agriculture-based climate mitigation strategies and highlights a key role for synthetic biology in mitigation pathways. Regular re-evaluation of strategies should be performed to ensure that they are meaningful for climate change mitigation as other factors evolve.
|
|
Scooped by
Jean-Michel Ané
November 30, 10:02 AM
|
Biological nitrogen fixation (BNF), which is catalyzed by a large nitrogenase enzyme complex, has evolved in both bacteria and archaea. Indeed, nitrogen-fixing species are found in diverse living environments, and BNF has evolved even in aerobic bacteria, although the function of nitrogenase is inhibited by oxygen. BNF is, however, highly energy-costing, requiring 16 ATPs in a single nitrogen fixation reaction. To explain this paradox, we hypothesized that nitrogen-fixing species gain not only nitrogen-fixing (nif) genes but also non-nif genes to facilitate nitrogen fixation. We examined over 3500 nitrogen-fixing genomes and found that they have gained genes directly or indirectly related to BNF in diverse types of living environments, so that nitrogen-fixing species tend to have larger genomes than their non-nitrogen-fixing relatives. Interestingly, the non-nif genes gained tend to be located near nif-gene clusters, probably to achieve proximity effects such as coordinated gene regulation. For example, the most frequent among the genes gained are ABC transporter genes, which facilitate the absorption and physiological metabolism of carbon (e.g., sugars), nitrogen (e.g., amino acids), and trace elements (e.g., molybdenum), and many ABC transporter genes lie close to nif-gene clusters. From our findings, we propose the following scenario: BNF evolved in many archaea and bacteria because BNF is advantageous to its hosts, although it incurs a high energy cost. Then, gaining genes to facilitate BNF compensates the cost of BNF, facilitating the spread of nitrogen fixers to all living habitats. This expansion benefits the biosphere, as nitrogen is essential for all organisms.
|
|
Scooped by
Jean-Michel Ané
November 25, 9:56 AM
|
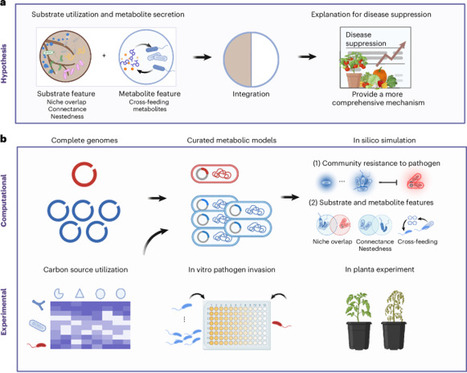
Understanding how plant-associated microbiomes resist phytopathogen invasion remains a key challenge in natural ecosystems. Here we combined genome-scale metabolic models with synthetic community experiments, both in vitro and in planta, to unravel the mechanisms driving pathogen suppression. We developed curated genome-scale models for each strain, incorporating 48 common resource utilization profiles to fully capture their metabolic capacities. Trophic interactions inferred from models effectively predicted pathogen invasion outcomes across diverse microbial communities and nutrient environments. Importantly, considering both substrate and metabolite features provided a more holistic understanding of pathogen suppression. In particular, cross-feeding metabolites within the native community emerged as crucial yet often overlooked predictors of community resistance, disproportionally favouring native species over invaders. This study lays the foundation for designing disease-resistant microbiomes, with broad implications for mitigating pathogen exposure in diverse environments.
|
|
Scooped by
Jean-Michel Ané
November 24, 6:04 PM
|
Background and aims
Intercropping has been demonstrated to enhance crop productivity and phosphorus (P) uptake, in which root-microbe interactions played crucial roles. Our previous results showed that this beneficial effect of intercropping depends largely on a match between root traits and rhizosphere processes. However, little is known about the role of soil microbial communities in underground processes.
Methods
Using a long-term field experiment with three P-fertilizer application rates and five cropping systems of maize, we integrated crop productivity, root physiological traits, root morphological traits and microbial amplicon sequencing data.
Results
Our findings revealed that intercropping significantly enhanced maize crop productivity and P uptake, accompanied by increased plasticity of morphological and physiological root traits compared with monoculture. Additionally, intercropping with different companion crops significantly altered the soil microbial community structure of maize, while P-fertilizer application rates had minimal effect. Network analysis showed that intercropping promoted more complex and stable microbial networks characterized with increased cooperative relationships, relative to monoculture. The relative abundance of keystones enriched in intercropping systems were positively correlated with crop productivity and P uptake, explaining 41.11% of the variation in maize grain yield. Structural equation modeling (SEM) further indicated that keystones enhanced grain yields of maize by inducing carboxylate secretion and fostering more acquisitive root morphological traits.
Conclusion
Our study elucidates how intercropping optimizes root-microbe synergies to improve P efficiency, highlighting the potential for targeted manipulation of keystone microbial taxa to enhance P acquisition and crop performance in sustainable agricultural systems.
|
|
Scooped by
Jean-Michel Ané
November 24, 1:19 PM
|
Legume root nodulation with nitrogen-fixing bacteria requires precise control via root-shoot-root autoregulation of nodulation (AON). Post-translationally modified root-derived CLAVATA3/Embryo Surrounding Region-Related (CLE) peptides signal through shoot acting leucine-rich repeat receptors (CLAVATAs) to regulate nodule number and this pathway is a target to optimise nodulation. We characterise the AON system in the crop model pea (Pisum sativum L.) and address key gaps in our understanding of AON; the role of parallel signalling pathways, shoot receptor complexes and downstream targets. We use novel mutant combinations, overexpression, grafting, gene expression and careful analysis of infection and nodule organogenesis using GFP-labelled rhizobium. These studies provide evidence that in pea both PsCLE12 and PsCLE13 require arabinosylation via PsRDN1. Perception of PsCLE12 and PsCLE13 in the shoot to suppress the mature nodules in the root requires the pea CLAVATA1 orthologue PsNARK and PsCLV2. However, we found little evidence that PsCLE12 and PsCLE13 suppress infection thread development or that they act via PsTML1 and/or PsTML2, root acting suppressors of nodulation, indicating a role for additional CLE signals. Grafting and double mutant studies indicate that PsNARK can act together with PsCLV2, but also independently, to influences nodulation providing in planta evidence for shoot receptor complexes that control AON.
|



















Outstanding preprint from @leocastanedo.bsky.social, @katharinamel1.bsky.social, @pierremarcdelaux.bsky.social and coll. on an evolutionary conserved module for intracellular symbiosis.