Your new post is loading...

|
Scooped by
?
September 9, 12:53 PM
|
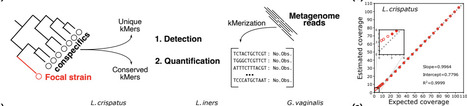
Motivation Accurate detection and quantification of bacterial strains in clinical samples is necessary to measure their colonization and persistence. Past methods to achieve this relied either on strain-specific qPCR assays, or shotgun metagenomic read mapping approaches. The resident microbial community is a major source of interference in both assays because it can contain conspecific strains bearing similarity to the focal strain(s). Results We present kSanity, a k-mer based application for the detection and quantification of targeted bacterial strains in shotgun metagenomic data. Because kSanity uses exact string matches between the reads and reference, it is less sensitive to inter- ference by conspecific strains. We test the performance of kSanity using a combination of in silico spike-in experiments, and in vivo observational data. Our results demonstrate that kSanity provides precise and accurate quantification of targeted bacterial strains, even when they are present at low sequence coverage in the metagenome. Availability and implementation kSanity is available at: https://github.com/ravel-lab/kSanity
|
|
Scooped by
?
September 9, 12:25 PM
|
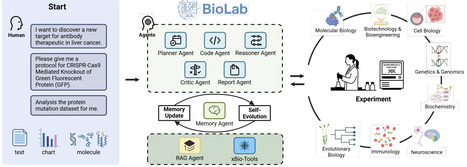
Scientific discovery in the life sciences remains hindered by fragmented workflows, narrow-scope computational models, and inefficient links between in silico prediction and wet-lab validation. We present BioLab, a multi-agent system that integrates domain-specialized foundation models to automate end-to-end biological research. BioLab comprises eight collaborating agents, including a Planner, Reasoner, and Critic, orchestrated through a Memory Agent that enables iterative refinement via retrieval-augmented generation and a suite of 219 computational xBio-Tools spanning five biological scales (DNA, RNA, protein, cell, and chemical). These tools are built on the xTrimo Universe, a collection of 104 models derived from six foundation models (xTrimoChem, Protein, RNA, DNA, Cell, and Text), the majority of which achieve state-of-the-art (91-100% SOTA ratios) on domain benchmarks. Across standard reasoning tasks (PubMedQA, MMLU-Pro/Biology, GPQA-diamond), BioLab consistently outperformed leading large language models, including GPT-4o, Gemini-2.5, and DeepSeek-R1. Beyond benchmarks, BioLab autonomously executed a fully computational pipeline for de novo macrophage-targeting antibody design, progressing from target mining to multi-objective antibody optimization, where molecular dynamics simulations revealed structural mechanisms underlying enhanced affinity of optimized variants. Closing the computational-experimental loop, BioLab designed optimized antibodies (Pem-MOO-1, Pem-MOO-2) that achieved IC50 values of 0.01-0.016 nM, markedly surpassing the parental Pembrolizumab ( 0.027 nM) for PD-1. Functional assays confirmed enhanced pathway blockade and improved multi-parameter performance profiles. Together, these results establish BioLab as a generalizable framework for AI-native scientific discovery, demonstrating how multi-agent systems coupled with foundation models can autonomously generate, execute, and experimentally validate novel biological hypotheses.
|
|
Scooped by
?
September 9, 12:08 PM
|
Iron plaque (IP) on rice root surfaces has been extensively documented as a natural barrier that effectively reduces contaminant bioavailability and accumulation. However, its regulatory mechanisms in rhizospheric methane oxidation and biological nitrogen fixation (BNF) remain elusive. This study reveals a previously unrecognized function of IP: mediating methanotrophic nitrogen fixation through coupled aerobic methane oxidation and IP reduction (Fe-MOX). Using a hydroponic coculture system integrating methane-oxidizing bacteria and rice seedlings, we demonstrated that IP enhanced microbial methane oxidation by 46.8% and significantly stimulated BNF rate by 33.6%, with methane-derived carbon accounting for 89.1% of the BNF energy source. Notably, dissolved iron removal did not diminish the BNF enhancement, excluding mediation by soluble iron species. Intriguingly, ferrihydrite supplementation at equivalent iron concentrations failed to replicate the BNF stimulation observed with IP, suggesting the indispensability of root-associated iron redox cycling. Mechanistic analyses identified that Methylosinus/ Methylocystis species mediated Fe(III) reduction, synergistically collaborating with specific rhizobial strains to execute Fe-MOX-dependent BNF. These findings uncover a previously overlooked yet pronounced contribution of IP to BNF, providing novel insights for developing dual-strategy approaches to mitigate methane emissions and reduce nitrogen fertilizer dependency in paddy ecosystems.
|
|
Scooped by
?
September 9, 11:49 AM
|
Synthetic biology often employs heterologous enzymatic reactions to reprogram cell metabolism or otherwise introduce novel functions. However, precise control of a particular metabolic pathway can be difficult to achieve because cofactors are shared with endogenous enzymes from a common pool. Recently, the use of noncanonical cofactors (NCCs) has emerged as a promising approach to bypass this problem by isolating desired reactions without the need for a physical barrier. Metabolic pathways that exclusively utilize NCCs can be insulated from the native machinery of the host cell, allowing them to function independently of the thermodynamic constraints imposed by sharing cofactors. This perspective explores the different types of NCCs and their synthesis methods, advancements in engineering NCC-dependent enzymes, and the potential applications of NCC-utilizing cells across various areas of synthetic biology.
|
|
Scooped by
?
September 9, 11:40 AM
|
The 5′ untranslated region (5′UTR) plays a crucial regulatory role in messenger RNA (mRNA), with modified 5′UTRs extensively utilized in vaccine production, gene therapy, etc. Nevertheless, manually optimizing 5′UTRs may encounter difficulties in balancing the effects of various cis-elements. Consequently, multiple 5′UTR libraries have been created, and machine learning models have been employed to analyze and predict translation efficiency (TE) and protein expression, providing insights into critical regulatory features. On the one hand, these screening libraries, based on TE and mean ribosome load, struggle to accurately quantify protein expression; on the other hand, a precise method for quantifying 5′UTRs necessitates a significantly costlier library. To resolve this dilemma, we constructed a library utilizing firefly luciferase as the reporter to measure accurate protein expression. In addition, we optimized the library construction method by clustering mRNA sequences to reduce redundant data and minimize the size of the dataset. This dual strategy by increasing accuracy and reducing dataset size was found to be effective in predicting the 5′UTRs from the PC3 cell line.
|
|
Scooped by
?
September 9, 10:58 AM
|
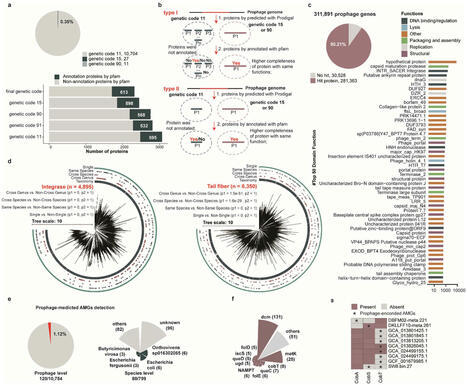
Prophages, viruses integrated into bacterial or archaeal genomes, can carry cargo that confers beneficial phenotypes to the host. The porcine gut microbiota constitutes a complex, dynamic, and interconnected ecosystem, yet the distribution of prophages and their unique functional characteristics within this microbial community remains poorly understood. In this study, we identified 10,742 prophage genomes through systematic screening of 7,524 prokaryotic genomes from porcine gut sources, representing both bacterial and archaeal lineages, with the distribution of integrated prophages exhibiting pronounced heterogeneity across host species. Additionally, 1.70% (183/10,742) of prophages exhibited a broad host range infectivity, while 5.07% (545/10,742) of integrated prophages enhanced prokaryotic adaptive immune capabilities by augmenting or directly providing host defense mechanisms. Notably, within tripartite phage-phage-host interactions network analysis, we observed that these prophages (n = 15) exhibit preferential acquisition of exogenous invasive phage sequences through CRISPR spacer integration mechanisms. Functional annotation revealed that prophage-encoded integrases and tail tube proteins may be critical determinants of phage host specificity. In addition, key auxiliary metabolic genes are encoded in the prophage of the pig intestinal tract, such as those promoting the synthesis of host microbiota-derived vitamin B12, encoded antibiotic resistance genes, and virulence factors that provide the host with a survival advantage. Furthermore, comparative analysis with existing viral and phage sequences uncovered a substantial reservoir of high-quality novel prophage sequences. Our findings systematically investigated the diversity of prophages in the pig gut, further characterizing their host range, functional attributes, and interactions with both host bacteria and other phages, through large-scale analysis of porcine gut microbiota genomes. This work offers new insights into the ecological roles of prophages and provides valuable genomic resources for studying prophages in this ecosystem.
|
|
Scooped by
?
September 9, 10:37 AM
|
Although dynamical systems models are a powerful tool for analysing microbial ecosystems, challenges in learning these models from complex microbiome datasets and interpreting their outputs limit use. We introduce the Microbial Dynamical Systems Inference Engine 2 (MDSINE2), a Bayesian method that learns compact and interpretable ecosystems-scale dynamical systems models from microbiome timeseries data. Microbial dynamics are modelled as stochastic processes driven by interaction modules, or groups of microbes with similar interaction structure and responses to perturbations, and additionally, noise characteristics of data are modelled. Our open-source software package provides multiple tools for interpreting learned models, including phylogeny/taxonomy of modules, and stability, interaction topology and keystoneness. To benchmark MDSINE2, we generated microbiome time-series data from two murine cohorts that received faecal transplants from human donors and were then subjected to dietary and antibiotic perturbations. MDSINE2 outperforms state-of-the-art methods and identifies interaction modules that provide insights into ecosystems-scale interactions in the gut microbiome. This Bayesian statistical method uses timeseries microbiome data to infer interaction modules and is tested using a faecal transplant experiment in mice.
|
|
Scooped by
?
September 9, 10:17 AM
|
The photoautotrophic lifestyle of cyanobacteria has to cope with the successive diurnal changes. We previously highlighted a unique function of the carbon control protein, SbtB, and its effector molecule c-di-AMP, for nighttime survival through the regulation of glycogen anabolism. However, the extent to which c-di-AMP and SbtB impact the cellular metabolism for day-night survivability remained elusive. To gain a better understanding, we compared the metabolomic and proteomic landscapes of ΔsbtB and the c-di-AMP-free (ΔdacA) mutants of Synechocystis sp. PCC 6803. While our results indicated that the cellular role of SbtB is restricted to carbon/glycogen metabolism, the diurnal lethality of ΔdacA seemed to be a sum of the dysregulation of multiple processes, including photosynthesis and redox regulation. Further, we showed an impact of c-di-AMP on central carbon/nitrogen metabolism, which is linked to NtcA transcription regulation and highlighted by an imbalance of glutamine/glutamate ratio as well as the reduction of arginine pathway metabolites. We further identified the HCO3− uptake systems, BicA and BCT1, as novel SbtB targets, in agreement with its broader role in regulating carbon homeostasis.
|
|
Scooped by
?
September 9, 10:13 AM
|
Plastic pollution is a major environmental challenge, with millions of tonnes produced annually and accumulating in ecosystems, causing long-term harm. Conventional disposal methods, such as landfilling and incineration, are often inadequate, emphasising the need for sustainable solutions like bioremediation. However, the bacterial biodiversity involved in plastic biodegradation remains poorly understood. To address this gap, we present the Plastic-Microbial BioRemediation (Plastic-MBR) database, a curated multi-omics resource that integrates publicly available genetic and enzymatic data related to putative plastic-degrading microorganisms. This database supports in silico analyses of metagenomic data from plastic-contaminated environments and comparative genomics, aiming to identify microbial taxa with potential plastic-degrading functions. We validated the functionality of the Plastic-MBR database by applying it to metagenomic datasets from plastic-contaminated soil and river water, successfully identifying numerous putative plastic-degrading genes across diverse microbial taxa. These results support the use of the Plastic-MBR database as a tool to identify candidate bacteria for future experimental validation, strain isolation, and functional studies, ultimately contributing to a deeper understanding of microbial potential in plastic bioremediation. While this study focuses on database development and computational validation, future studies will be essential to confirm and translate these genomic predictions into effective bioremediation strategies.
|
|
Scooped by
?
September 9, 9:55 AM
|
Microbial influence on cancer development and therapeutic response is a growing area of cancer research. Although it is known that microorganisms can colonize certain tissues and contribute to tumor initiation, the use of deep sequencing technologies and computational pipelines has led to reports of multi-kingdom microbial communities in a growing list of cancer types. This has prompted discussions on the role and scope of microbial presence in cancer, while raising the possibility of microbiome-based diagnostic, prognostic and therapeutic tools. However, additional investigation and thorough validation of cancer microbiome findings are required before this translational potential can be realized. Here we provide historical context and a conceptual framework for the so-called cancer microbiome and summarize experimental studies into tumor-associated bacteria, fungi and other microorganisms. We also discuss the current evidence for microbial colonization of tumors and their varied influence on the disease, including recent debates. Finally, we consider outstanding questions and discuss our outlook for the field. This Review discusses what comprises the ‘cancer microbiome’, summarizing the studies on tumour-associated microbes, examining the evidence and assessing their impact on the disease.
|
|
Scooped by
?
September 9, 9:26 AM
|
Plant roots interact with pathogenic and beneficial microbes in the soil. While root defense barriers block pathogens, their roles in facilitating beneficial plant–microbe associations are understudied. Here, we examined the impact of specific root defense barriers on the well-known beneficial association between Arabidopsis thaliana and the plant growth-promoting rhizobacterium Pseudomonas simiae WCS417. Using 15 Arabidopsis mutants with alterations in structural (cutin, suberin, callose, and lignin) and chemical (camalexin and glucosinolates) defense barriers, we demonstrate that some barriers impact WCS417-mediated plant growth responses and its root colonization. Root exudates from Arabidopsis wild-type (WT) and mutant plants differentially affected the WCS417 transcriptome, with camalexin notably impacting bacterial motility and chemotaxis, which was also confirmed by in vitro studies. On the plant side, WCS417-induced transcriptome changes in the roots of defense barrier mutants were significantly different from those in WT plants, particularly affecting growth and defense-related processes. Specifically, the data indicated altered activity of reactive oxygen species in several of the defense barrier mutants, which was confirmed in planta. Our data suggest that various root defense barriers play a role in balancing growth and defense during this mutualistic interaction, thereby impacting the establishment and effectiveness of plant mutualists, extending their established role in disease resistance.
|
|
Scooped by
?
September 9, 1:07 AM
|
Prokaryotic genomes are gene-dense, so genes in the same orientation are often separated by short intergenic sequences or even overlap. Many mechanisms of regulation depend on open reading frames (ORFs) being spatially close to one another. Here, we describe one such mechanism, translational coupling, where translation of one gene promotes translation of a co-oriented neighboring gene. Translational coupling has been observed across the prokaryotic kingdom. Coupling is most efficient when the intergenic distance between ORFs is small. Coupling efficiency is influenced by RNA secondary structure, the presence of a Shine-Dalgarno (SD) sequence, and potentially by other cis-acting elements. While the mechanism of translational coupling has not been firmly established, two models have been proposed. In the RNA unfolding model, translation of the upstream gene in a pair disrupts inhibitory RNA secondary structure around the start codon of the downstream gene. Alternatively, the reinitiation model proposes that the same ribosome—either the 30S or complete 70S—translates both genes in a coupled pair. We describe evidence in support of each model, and we discuss the functional implications of translational coupling.
|
|
Scooped by
?
September 9, 12:31 AM
|
Dinitrogen (N2) fixation provides bioavailable nitrogen to the biosphere. However, in some habitats (e.g., sediments), the metabolic pathways of organisms carrying out N2 fixation are unclear. We present metabolic models representing various chemotrophic N2 fixers, which simulate potential pathways of electron transport and energy flow, resulting in predictions of whole-cell stoichiometries. By balancing mass, electrons, and energy for metabolic half-reactions, we quantify the electron usage for nine N2 fixers. Our results demonstrate that all modeled organisms fix sufficient N2 for growth. Aerobic organisms allocate more electrons to N2 fixation and growth, yielding more biomass and fixing more N2, while methanogens using acetate and organisms using sulfate allocate fewer electrons. This work can be applied to investigate the depth distribution of N2 fixers based on nutrient availability, complementing field measurements of biogeochemical processes and microbial communities.
|
|
|
Scooped by
?
September 9, 12:34 PM
|
Artificial intelligence (AI) models have been proposed for hypothesis generation, but testing their ability to drive high-impact research is challenging since an AI-generated hypothesis can take decades to validate. Here, we challenge the ability of a recently developed large language model (LLM)-based platform, AI co-scientist, to generate high-level hypotheses by posing a question that took years to resolve experimentally but remained unpublished: how could capsid-forming phage-inducible chromosomal islands (cf-PICIs) spread across bacterial species? Remarkably, the AI co-scientist’s top-ranked hypothesis matched our experimentally confirmed mechanism: cf-PICIs hijack diverse phage tails to expand their host range. We critically assess its five highest-ranked hypotheses, showing that some opened new research avenues in our laboratories. We benchmark its performance against other LLMs and outline best practices for integrating AI into scientific discovery. Our findings suggest that AI can act not just as a tool but as a creative engine, accelerating discovery and reshaping how we generate and test scientific hypotheses.
|
|
Scooped by
?
September 9, 12:19 PM
|
Guided by this conceptual framework rooted in large language models, our study integrates DeepPGDB https://www.deeppgdb.chat model fine-tuning with prompt engineering to construct the first AI-powered plant genomics database. DeepPGDB integrates bioinformatics tools through model integration, combined with fine-tuning, prompt engineering, and retrieval-augmented generation (RAG). This enables an AI served as a scheduler to accurately identify user intent and translate it into standardized tool invocation command. DeepPGDB determines tasks from two perspectives: task type and data type.
|
|
Scooped by
?
September 9, 11:54 AM
|
Pectinases are indispensable biocatalysts for pectin degradation in food and bioprocessing industries, yet natural enzymes often lack tailored functionalities for modern applications. While a previous review discussed pectinases in terms of production and application, this review particularly discusses an integrated framework for robust pectinases. This framework combines enzyme mining, protein engineering, and AI-assisted design to systematically discover, optimize, and customize pectinases. These synergistic strategies, in fact, have been widely explored in recent years to enable precise development of biocatalysts with enhanced industrial traits, moving beyond traditional single-approach-based enzyme improvement. Specifically, we discuss how cutting-edge methodologies, such as data-driven discovery and intelligent protein engineering, accelerate robust pectinase development, while emerging purification and bioprocessing techniques expand their applications in juice/wine production, textile bioscouring, and agricultural waste valorization. By unifying novel microbial sources, mechanistic insights, and engineering advances, these holistic approaches offer transformative potential for biocatalyst development, including pectinases. In this way, this review consolidates recent progress to guide next-generation pectinase development through combinatorial biotechnology, providing actionable insights for advancing sustainable industrial processes.
|
|
Scooped by
?
September 9, 11:45 AM
|
Human Bone Morphogenetic Protein-2 (hBMP-2) serves as a critical regulator in bone and cartilage formation; however, its industrial application is hindered by its inherent tendency to form inclusion bodies in prokaryotic expression systems. To address this issue, we established a recombinant hBMP-2 (rhBMP-2) expression system using the pCold II plasmid and the SHuffle T7 strain. We explored several strategies to enhance the solubility of rhBMP-2, including coexpression with molecular chaperones, vesicle-mediated secretory expression, fusion expression with synthetic intrinsically disordered proteins (SynIDPs), and fusion expression with small-molecule peptide tags. Our results showed that coexpression with the molecular chaperone pGro7 significantly improved the solubility of rhBMP-2. Fusion with SynIDPs led to complete solubility of rhBMP-2; however, the protein was expressed exclusively in the monomeric form. Among the tested small-molecule peptide tags, GB1 was the most effective, achieving fully soluble rhBMP-2 expression. Western blot analysis confirmed the coexistence of monomeric and dimeric forms of rhBMP-2. Subsequent purification of rhBMP-2 through metal chelate chromatography resulted in an expression level of 109.7 ± 5.0 mg·L–1. In summary, we successfully demonstrated fully soluble expression of rhBMP-2 in Escherichia coli, providing a valuable foundation for its industrial-scale production.
|
|
Scooped by
?
September 9, 11:20 AM
|
Tandem repetition is one of the major processes underlying genome evolution and phenotypic diversification. While newly formed tandem repeats are often easy to identify, it is more challenging to detect repeat copies as they diverge over evolutionary timescales. Existing programs for finding tandem repeats return markedly different results, and it is unclear which predictions are more correct and how much room remains for improvement. Here, we introduce DetectRepeats, a new method that uses empirical information about structural repeats to improve the accuracy of repeat detection. We show that DetectRepeats advances the state-of-the-art by finding highly divergent repeats with relatively few false positive detections. We apply DetectRepeats to genomes across the tree of life to discover an enrichment of detectable tandem repeats within different genes, genome regions, and taxa. Furthermore, we use phylogenetic reconciliation to determine that some tandem repeats continue to evolve through intra-repeat unit replacement. In this manner, tandem repeats serve as a renewable genetic resource offering a bountiful source of alternative genetic material. Our work unlocks the confident detection of ancient tandem repeats, opening a doorway to future discoveries. DetectRepeats is part of the DECIPHER package for the R programming language and available via Bioconductor.
|
|
Scooped by
?
September 9, 10:52 AM
|
Protein design is undergoing a revolution driven by artificial intelligence (AI), transforming how we engineer proteins for applications in drug discovery, biotechnology and synthetic biology. By navigating the immense complexity of protein sequence space and overcoming the limitations of structural and functional data, AI enables unprecedented precision and speed in designing novel proteins with tailored functions. Central to this Review is a comprehensive and actionable roadmap for designers, providing step-by-step guidance on how to integrate state-of-the-art AI tools into protein design workflows, including tools for structural and functional prediction as well as generative models for de novo design. To illustrate this roadmap in practice, we present case studies showcasing AI-driven protein design, from engineering therapeutic proteins to designing novel proteins that unlock enzyme functions and reprogramme biomolecular systems. Looking ahead, we outline future directions highlighting the vast potential of AI to revolutionize synthetic biology, expedite drug development and drive sustainable biotechnology, positioning it as a transformative force at the forefront of protein design. Artificial intelligence is revolutionizing protein design by enabling precise navigation of sequence space and accelerating the creation of functional proteins. In this Review, a practical roadmap guides integration of artificial intelligence tools into design workflows, highlighting structural prediction, generative models and case studies spanning engineering therapeutic proteins to designing new ones.
|
|
Scooped by
?
September 9, 10:35 AM
|
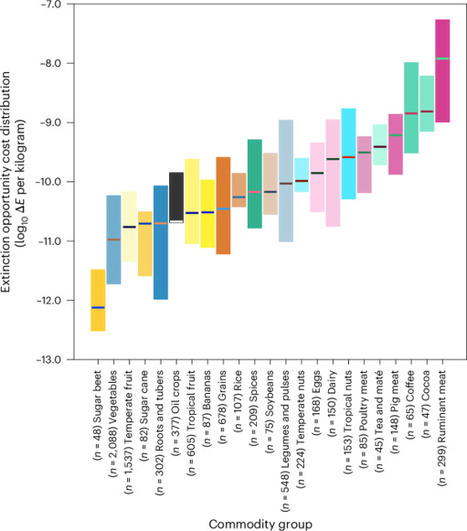
Agriculturally driven habitat degradation and destruction is the biggest threat to global biodiversity. Yet the impact of different foods and where they are produced on species extinction risks, and the mitigation potential of different interventions, remain poorly quantified. Here we link the LIFE biodiversity metric—a high-resolution global layer describing the marginal impact of land use on extinctions of ~30,000 vertebrate species—with food consumption and production data and provenance modelling. Using an opportunity cost framing, we estimate that the impact of producing 1 kg of different food commodities on species extinction risks varies widely both across and within foods, in many cases by more than an order of magnitude. Despite marked differences in per capita impacts across countries, there are consistent patterns that could be leveraged for mitigating harm to biodiversity. In particular, animal products and commodities grown in the tropics are generally much more impactful than staple crops and vegetables. What we eat, as well as where and how it is grown, impacts species extinction risks through agricultural land use. Using a new global biodiversity impact data product, this study estimates how many species extinctions may potentially be caused by the production and consumption of different food types on a country-by-country basis.
|
|
Scooped by
?
September 9, 10:15 AM
|
In mammals, blood sugar levels are tightly controlled by two hormones: insulin and glucagon. In flowering plants, a comparable regulatory mechanism exists, mediated by the sugar-signalling molecule trehalose 6-phosphate (Tre6P). Similar to insulin, Tre6P functions as a signal and negative feedback regulator of sucrose, the main transport sugar in vascular plants. In the model plant Arabidopsis thaliana and likely all other angiosperms, Tre6P is predominantly synthesized in the vasculature, an ideal position to integrate systemic sugar status with whole-plant developmental decision-making. Genes encoding components of Tre6P dephosphorylation and signalling show broader expression patterns suggesting movement and signalling of Tre6P outside the vasculature to coordinate plant metabolism and development.
|
|
Scooped by
?
September 9, 10:07 AM
|
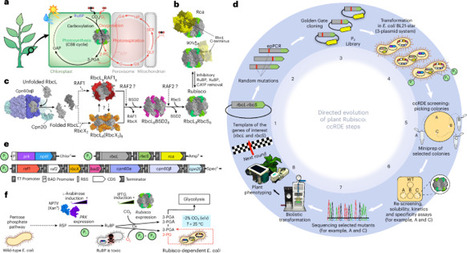
A new Escherichia coli laboratory evolution screen for detecting plant ribulose-1,5-bisphosphate carboxylase/oxygenase (Rubisco) mutations with enhanced CO2-fixation capacity has identified substitutions that can enhance plant productivity. Selected were a large subunit catalytic (Met-116-Leu) mutation that increases the kcat of varying plant Rubiscos by 25% to 40% and a solubility (Ala-242-Val) mutation that improves plant Rubisco biogenesis in E. coli 2- to 10-fold. Plastome transformation of either mutation into the tobacco plastome rbcL gene had no impact on leaf Rubisco production, photosynthesis or plant growth. However, tobacco transformed with low-abundance hybrid Arabidopsis Rubisco coding M116L improved plant exponential growth rate by ~75% relative to unmutated hybrid enzyme, with the A242V substitution increasing both hybrid Rubisco production and plant growth by ~50%. Our identification of mutations with the potential to enhance plant growth bodes well for broadening the survey of Rubisco sequence space for catalytic switches that can impart more substantive plant productivity improvements. This study demonstrates the power of directed evolution to unlock latent functional potential in plant Rubisco. By identifying mutations that enhance CO2 fixation and solubility, it advances avenues for improving crop photosynthesis and productivity.
|
|
Scooped by
?
September 9, 9:29 AM
|
Predicting small molecule binding sites on proteins remains a key challenge in structure-based drug discovery. While AlphaFold 3 has transformed protein structure prediction, accurate identification of functional sites such as ligand binding pockets remains a distinct and unresolved problem. Graph neural networks have emerged as promising tools for this task, but most current approaches focus on local structural features and are trained on relatively small datasets, limiting their ability to model long-range protein-ligand interactions. Here, we develop YuelPocket, a new graph neural network that addresses these limitations through an innovative global graph design featuring a virtual joint node connecting all protein residues and small molecule atoms to capture long-range interactions while maintaining linear computational complexity. Trained on the comprehensive MOAD dataset containing over 38,000 protein-small molecule complexes, YuelPocket achieves exceptional performance with AUC-ROC values of 0.85 and 0.89 on COACH420 and Holo4k datasets, respectively. docking
|
|
Scooped by
?
September 9, 1:14 AM
|
Metabolic engineering is rapidly evolving as a result of new advances in synthetic biology tools and automation platforms that enable high throughput strain construction, as well as the development of machine learning tools (ML) for biology. However, selecting genetic engineering targets that effectively guide the metabolic engineering process is still challenging. ML can provide predictive power for synthetic biology, but current technical limitations prevent the independent use of ML approaches without previous biological knowledge. Here, we present FluxRETAP, a simple and computationally inexpensive method that leverages the prior mechanistic knowledge embedded in genome-scale models for suggesting targets for genetic overexpression, downregulation or deletion, with the final goal of increasing the production of a desired metabolite. This method can provide a list of desirable engineering targets that can be combined with current ML pipelines. FluxRETAP captured 100% of reaction targets experimentally verified to improve Escherichia coli isoprenol production, 50% of targets that experimentally improved taxadiene production in E. coli and ∼60% of genetic targets from a verified minimal constrained cut-set in Pseudomonas putida, while providing additional high priority targets that could be tested. Overall, FluxRETAP is an efficient algorithm for identifying a prioritized list of testable genetic and reaction targets.
|
|
Scooped by
?
September 9, 1:01 AM
|
Genome-scale metabolic models (GEMs) are widely used in systems biology to investigate metabolism and predict perturbation responses. Automatic GEM reconstruction tools generate GEMs with different properties and predictive capacities for the same organism. Since different models can excel at different tasks, combining them can increase metabolic network certainty and enhance model performance. Here, we introduce GEMsembler, a Python package designed to compare cross-tool GEMs, track the origin of model features, and build consensus models containing any subset of the input models. GEMsembler provides comprehensive analysis functionality, including identification and visualization of biosynthesis pathways, growth assessment, and an agreement-based curation workflow. GEMsembler-curated consensus models built from four Lactiplantibacillus plantarum and Escherichia coli automatically reconstructed models outperform the gold-standard models in auxotrophy and gene essentiality predictions. Optimizing gene-protein-reaction (GPR) combinations from consensus models improves gene essentiality predictions, even in the manually curated gold-standard models. GEMsembler explains model performance by highlighting relevant metabolic pathways and GPR alternatives, informing experiments to resolve model uncertainty. Thus, GEMsembler facilitates building more accurate and biologically informed metabolic models for systems biology applications.
|