Your new post is loading...

|
Scooped by
mhryu@live.com
April 15, 2018 3:52 PM
|
Extensive research efforts have been dedicated to describing degradation of wood, which is a complex process; hence, microorganisms have evolved different enzymatic and non-enzymatic strategies to utilize this plentiful plant material. This review describes a number of fungal and bacterial organisms which have developed both competitive and mutualistic strategies for the decomposition of wood and to thrive in different ecological niches. Through the analysis of the enzymatic machinery engaged in wood degradation, it was possible to elucidate different strategies of wood decomposition which often depend on ecological niches inhabited by given organism. Moreover, a detailed description of low molecular weight compounds is presented, which gives these organisms not only an advantage in wood degradation processes, but seems rather to be a new evolutionatory alternative to enzymatic combustion. Through analysis of genomics and secretomic data, it was possible to underline the probable importance of certain wood-degrading enzymes produced by different fungal organisms, potentially giving them advantage in their ecological niches. The paper highlights different fungal strategies of wood degradation, which possibly correlates to the number of genes coding for secretory enzymes. Furthermore, investigation of the evolution of wood-degrading organisms has been described.
|
|
Scooped by
mhryu@live.com
April 15, 2018 3:43 PM
|
Polyploidy, or whole-genome duplication (WGD), is usually an evolutionary dead end. Although polyploidy is a frequent and recurrent phenomenon, the number of WGDs that have become established in the long term is low. The occurrence of WGDs in the tree of life is not random and seems to correlate with periods of environmental upheaval. WGDs increase the adaptive potential of cells and organisms exposed to stressful conditions. The biased retention of genes following WGDs offers a unique evolutionary potential to evolve key innovations and to increase biological complexity in the long term. In cancer, WGD is a transient state that promotes aneuploidy, and is responsible for increased genetic variation and subsequent adaptive potential.
|
|
Scooped by
mhryu@live.com
April 15, 2018 3:35 PM
|
Bayesian methods have become very popular in molecular phylogenetics due to the availability of user-friendly software for running sophisticated models of evolution. However, Bayesian phylogenetic models are complex, and analyses are often carried out using default settings, which may not be appropriate. Here we summarize the major features of Bayesian phylogenetic inference and discuss Bayesian computation using Markov chain Monte Carlo (MCMC) sampling, the diagnosis of an MCMC run, and ways of summarizing the MCMC sample. We discuss the specification of the prior, the choice of the substitution model and partitioning of the data. Finally, we provide a list of common Bayesian phylogenetic software packages and recommend appropriate applications.
|
|
Scooped by
mhryu@live.com
April 15, 2018 3:25 PM
|
Although many organisms capture or respond to sunlight, few enzymes are known to be driven by light. Among these are DNA photolyases and the photosynthetic reaction centers. Here, we show that the microalga Chlorella variabilis NC64A harbors a photoenzyme that acts in lipid metabolism. This enzyme belongs to an algae-specific clade of the glucose-methanol-choline oxidoreductase family and catalyzes the decarboxylation of free fatty acids to n-alkanes or -alkenes in response to blue light. Crystal structure of the protein reveals a fatty acid–binding site in a hydrophobic tunnel leading to the light-capturing flavin adenine dinucleotide (FAD) cofactor. The decarboxylation is initiated through electron abstraction from the fatty acid by the photoexcited FAD with a quantum yield >80%. This photoenzyme, which we name fatty acid photodecarboxylase, may be useful in light-driven, bio-based production of hydrocarbons.
|
|
Scooped by
mhryu@live.com
April 15, 2018 10:44 AM
|
Living organisms have evolved mechanisms for adjusting their metabolism to adapt to environmental nutrient availability. Terrestrial animals utilize the ornithine–urea cycle to dispose of excess nitrogen derived from dietary protein. Here, we identified an active ornithine–ammonia cycle (OAC) in cyanobacteria through an approach combining dynamic 15N and 13C tracers, metabolomics, and mathematical modeling. The pathway starts with carbamoyl phosphate synthesis by the bacterial- and plant-type glutamine-dependent enzyme and ends with conversion of arginine to ornithine and ammonia by a novel arginine dihydrolase. An arginine dihydrolase–deficient mutant showed disruption of OAC and severely impaired cell growth when nitrogen availability oscillated. We demonstrated that the OAC allows for rapid remobilization of nitrogen reserves under starvation and a high rate of nitrogen assimilation and storage after the nutrient becomes available. Thus, the OAC serves as a conduit in the nitrogen storage-and-remobilization machinery in cyanobacteria and enables cellular adaptation to nitrogen fluctuations.
|
|
Scooped by
mhryu@live.com
April 15, 2018 10:21 AM
|
In two-component systems, a sensor histidine kinase undergoes autophosphorylation and transfers a phosphate group to its cognate response regulator, which then mediates cellular responses by binding to DNA, performing enzymatic reactions, or interacting with other proteins. In the FixL-FixJ two-component system of the plant root nodule symbiont Bradyrhizobium japonicum , the histidine kinase FixL undergoes autophosphorylation only when it is not bound to oxygen. This ensures that its cognate response regulator FixJ stimulates the expression of genes required for nitrogen fixation only under low-oxygen conditions. Wright et al . combined high- and low-resolution structural analyses with modeling techniques and functional analysis to generate a model of signal relay through the FixL-FixJ two-component system. The model shows how the dissociation of oxygen from FixL stimulates FixL autophosphorylation and phosphotransfer from FixL to FixJ.
|
|
Scooped by
mhryu@live.com
April 13, 2018 11:38 AM
|
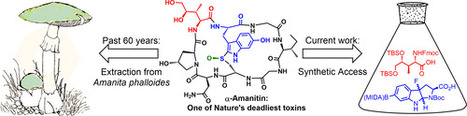
α-Amanitin is an extremely toxic bicyclic octapeptide isolated from the death-cap mushroom, Amanita phalloides. As a potent inhibitor of RNA polymerase II, α-amanitin is toxic to eukaryotic cells. Recent interest in α-amanitin arises from its promise as a payload for antibody–drug conjugates. For over 60 years, A. phalloides has been the only source of α-amanitin. Here we report a synthesis of α-amanitin, which surmounts the key challenges for installing the 6-hydroxy-tryptathionine sulfoxide bridge, enantioselective synthesis of (2S,3R,4R)-4,5-dihydroxy-isoleucine, and diastereoselective sulfoxidation.
|
|
Scooped by
mhryu@live.com
April 12, 2018 10:01 PM
|
Most genome-editing nucleases introduce a double-strand break at a target site, leaving compatible DNA ends that are repaired by the nonhomologous end joining repair pathway to regenerate the target site. This leads to a cycle of cleavage, target site regeneration by ligation, and subsequent recleavage by the nuclease. Here, we bypass this cycle by developing and testing an RNA-guided dual active site nuclease that introduces two noncompatible DNA breaks at a target site, deleting the majority of the target site so that it cannot be regenerated. The dual nuclease (TevCas9) robustly functions in HEK293 and biases genome-editing outcomes toward defined length deletions at high frequencies. Potential applications include directional oligonucleotide ligation, deletion of DNA-binding sites, and in-frame protein deletions.
|
|
Scooped by
mhryu@live.com
April 11, 2018 9:28 PM
|
Processing of double‐stranded RNA precursors into small RNAs is an essential regulator of gene expression in plant development and stress response. Small RNA processing requires the combined activity of a functionally diverse group of molecular components. However, in most of the plant species, there are insufficient mutant resources to functionally characterize each encoding gene. Here, mutations in loci encoding protein machinery involved in small RNA processing in soya bean and Medicago truncatula were generated using the CRISPR/Cas9 and TAL‐effector nuclease (TALEN) mutagenesis platforms. An efficient CRISPR/Cas9 reagent was used to create a bi‐allelic double mutant for the two soya bean paralogous Double‐stranded RNA‐binding2 (GmDrb2a and GmDrb2b) genes. These mutations, along with a CRISPR/Cas9‐generated mutation of the M. truncatula Hua enhancer1 (MtHen1) gene, were determined to be germ‐line transmissible. Furthermore, TALENs were used to generate a mutation within the soya bean Dicer‐like2 gene. CRISPR/Cas9 mutagenesis of the soya bean Dicer‐like3 gene and the GmHen1a gene was observed in the T0 generation, but these mutations failed to transmit to the T1 generation. The irregular transmission of induced mutations and the corresponding transgenes was investigated by whole‐genome sequencing to reveal a spectrum of non‐germ‐line‐targeted mutations and multiple transgene insertion events. Finally, a suite of combinatorial mutant plants were generated by combining the previously reported Gmdcl1a, Gmdcl1b and Gmdcl4b mutants with the Gmdrb2ab double mutant. Altogether, this study demonstrates the synergistic use of different genome engineering platforms to generate a collection of useful mutant plant lines for future study of small RNA processing in legume crops.
|
|
Scooped by
mhryu@live.com
April 11, 2018 12:20 AM
|
Culture-independent microbiome studies have increased our understanding of the complexity and metabolic potential of microbial communities. However, to understand the contribution of individual microbiome members to community functions, it is important to determine which bacteria are actively replicating. We developed an algorithm, iRep, that uses draft-quality genome sequences and single time-point metagenome sequencing to infer microbial population replication rates. The algorithm calculates an index of replication (iRep) based on the sequencing coverage trend that results from bi-directional genome replication from a single origin of replication. We apply this method to show that microbial replication rates increase after antibiotic administration in human infants. We also show that uncultivated, groundwater-associated, Candidate Phyla Radiation bacteria only rarely replicate quickly in subsurface communities undergoing substantial changes in geochemistry. Our method can be applied to any genome-resolved microbiome study to track organism responses to varying conditions, identify actively growing populations and measure replication rates for use in modeling studies.
|
|
Scooped by
mhryu@live.com
April 10, 2018 11:58 PM
|
pSC101 is a narrow host range, low-copy plasmid commonly used for genetically manipulating Escherichia coli. As a byproduct of a genetic screen for a more sensitive lactam biosensor, we identified multiple novel mutations that increase the copy number of plasmids with the pSC101 origin. All mutations identified in this study occurred on plasmids which also contained at least one mutation localized to the RepA protein encoded within the origin. Homology modelling predicts that many of these mutations occur within the dimerization interface of RepA. Mutant RepA resulted in plasmid copy numbers between ~31 and ~113 copies/cell, relative to ~5 copies/cell in wild-type pSC101 plasmids. Combining the mutations that were predicted to disrupt multiple contacts on the dimerization interface resulted in copy numbers of ~500 copies/cell, while also attenuating growth in host strains. Fluorescent protein production expressed from an arabinose-inducible promoter on mutant origin derived plasmids did correlate with copy number. Plasmids harboring RepA with one of two mutations, E83K and N99D, resulted in fluorescent protein production similar to that from p15a- (~20 copies/cell) and ColE1- (~31 copies/cell) based plasmids, respectively. The mutant copy number variants retained compatibility with p15a, pBBR, and ColE1 origins of replication. These pSC101 variants may be useful in future metabolic engineering efforts that require medium or high-copy vectors compatible with p15a- and ColE1-based plasmids.
|
|
Scooped by
mhryu@live.com
April 10, 2018 12:25 AM
|
In two important reports in this issue of PLoS Genetics, Sommer and colleagues convert much recent speculation into substance. In the first report [6], the Chandler and Sommer laboratories collaborate to explore the mechanism of transposition of element ISDra2. This transposon is a member of a family of elements that transpose via single-stranded DNA intermediates. Transposition is activated by irradiation of Deinococcus. The work not only documents the transposition mechanism, it reinforces the proposition that extensive lengths of single-stranded genomic DNA are generated in the early stages of genome reconstitution in this bacterium. As a bonus, the work provides hope for the development of in vivo transposition as a tool for genetic manipulation of this genome.
|
|
Scooped by
mhryu@live.com
April 10, 2018 12:14 AM
|
mRNAs can fold into complex structures that regulate gene expression. Resolving such structures de novo has remained challenging and has limited our understanding of the prevalence and functions of mRNA structure. We use SHAPE-MaP experiments in living E. coli cells to derive quantitative, nucleotide-resolution structure models for 194 endogenous transcripts encompassing approximately 400 genes. Individual mRNAs have exceptionally diverse architectures, and most contain well-defined structures. Active translation destabilizes mRNA structure in cells. Nevertheless, mRNA structure remains similar between in-cell and cell-free environments, indicating broad potential for structure-mediated gene regulation. We find that the translation efficiency of endogenous genes is regulated by unfolding kinetics of structures overlapping the ribosome binding site. We discover conserved structured elements in 35% of UTRs, several of which we validate as novel protein binding motifs. RNA structure regulates every gene studied here in a meaningful way, implying that most functional structures remain to be discovered.
|
|
|
Scooped by
mhryu@live.com
April 15, 2018 3:48 PM
|
Horizontal transfer, gene loss, and duplication result in dynamic bacterial genomes shaped by a complex mixture of different modes of evolution. Closely related strains can differ in the presence or absence of many genes, and the total number of distinct genes found in a set of related isolates—the pan-genome—is often many times larger than the genome of individual isolates. We have developed a pipeline that efficiently identifies orthologous gene clusters in the pan-genome. This pipeline is coupled to a powerful yet easy-to-use web-based visualization for interactive exploration of the pan-genome. The visualization consists of connected components that allow rapid filtering and searching of genes and inspection of their evolutionary history. For each gene cluster, panX displays an alignment, a phylogenetic tree, maps mutations within that cluster to the branches of the tree and infers gain and loss of genes on the core-genome phylogeny. PanX is available at pangenome.de. Custom pan-genomes can be visualized either using a web server or by serving panX locally as a browser-based application.
|
|
Scooped by
mhryu@live.com
April 15, 2018 3:40 PM
|
•A wealth of enzymes in industrial use are sourced from fungi. •Only a few fungal clades have been explored for metabolic engineering applications. •Novel biomass-degrading functions are likely to be found in the basal clades. •Anaerobic fungi have the highest diversity of biomass-degrading enzymes across fungi. •Modern biotechnology promises to unlock the potential of non-model fungi.
|
|
Scooped by
mhryu@live.com
April 15, 2018 3:33 PM
|
For several billion years, microorganisms and the genes they carry have mainly been moved by physical forces such as air and water currents. These forces generated biogeographic patterns for microorganisms that are similar to those of animals and plants ( 1 ). In the past 100 years, humans have changed these dynamics by transporting large numbers of cells to new locations through waste disposal, tourism, and global transport and by modifying selection pressures at those locations. As a consequence, we are in the midst of a substantial alteration to microbial biogeography. This has the potential to change ecosystem services and biogeochemistry in unpredictable ways.
|
|
Scooped by
mhryu@live.com
April 15, 2018 10:58 AM
|
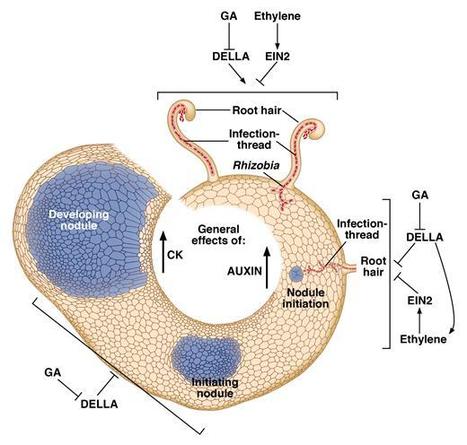
The role of gibberellin (GA) in legume nodulation is controversial, being reported to both enhance and inhibit the process. Now, in an elegant, multi-faceted investigation on peas McAdam et al. (2018)clearly show that GA is needed for nodule formation but inhibits the initiation of infection by Rhizobium. The investigators used genotypes varying in GA biosynthesis and signal transduction, combined with GA and GA-biosynthesis inhibitor applications, as well as an ethylene receptor mutation. Extensive measurements include developmental morphology and anatomy, gene expression, hormone levels and nitrogen fixation.
|
|
Scooped by
mhryu@live.com
April 15, 2018 10:31 AM
|
Recently, we published the first large-scale analysis of data from the Earth Microbiome Project (EMP) (1, 2), a truly multidisciplinary research program involving more than 500 scientists and 27,751 samples acquired from 43 countries. These samples represent myriad specimen types and span a wide range of biotic and abiotic factors, geographic locations, and physicochemical properties. The database (https://qiita.ucsd.edu/emp/) is still growing, with more than 100,000 amplicon data sets and more than 500 paired metagenomic and metabolomic data sets. Importantly, the techniques, data, and analytical tools are all standardized and publicly accessible, providing a framework to support research at a scale of integration that just 7 years ago seemed impossible.
|
|
Scooped by
mhryu@live.com
April 13, 2018 12:58 PM
|
Diazotrophic Bradyrhizobium spp. are well known for their ability to trigger nodule formation on a variety of legume species. In nodules, Bradyrhizobium utilizes plant-derived carbohydrates in exchange for fixed nitrogen. The genes essential for the nodulation and nitrogen-fixation trait are clustered in a genomic region, which is known as the ‘symbiotic island’. Recently, novel non-diazotrophic Bradyrhizobium spp. have been found to be highly abundant in soils, suggesting that these species can also have a ‘free-living’ life history. However, whether non-diazotrophic Bradyrhizobium spp. can live in association with plants remains elusive. In this study, we show that Bradyrhizobium spp. are common root endophytes of non-legume plant species – including Arabidopsis thaliana (Arabidopsis) – grown in an ecological setting. From a single Arabidopsis root, four Bradyrhizobium sp. strains (designated MOS001 to MOS004) were isolated. Comparative genome analysis revealed that these strains were genetically and functionally highly diverse, but did not harbour the nodulation and the nitrogen fixation gene clusters. Comparative colonization experiments, with MOS strains and nitrogen-fixing symbiotic strains, revealed that all tested Bradyrhizobium spp. can colonize the root endophytic compartment of Arabidopsis. This study provides evidence that both diazotrophic and non-diazotrophic Bradyrhizobium spp. colonize the root endophytic compartment of a wide variety of plant species, including the model species Arabidopsis. This demonstrates that plant roots form a major ecological niche for Bradyrhizobium spp., which might be ancestral to the evolution of the nodulation and nitrogen-fixation trait in this genus.
|
|
Scooped by
mhryu@live.com
April 13, 2018 1:31 AM
|
Elimination or mitigation of the toxic effects of chemical waste released to the environment by industrial and urban activities relies largely on the catalytic activities of microorganisms—specifically bacteria. Given their capacity to evolve rapidly, they have the biochemical power to tackle a large number of molecules mobilized from their geological repositories through human action (e.g., hydrocarbons, heavy metals) or generated through chemical synthesis (e.g., xenobiotic compounds). Whereas naturally occurring microbes already have considerable ability to remove many environmental pollutants with no external intervention, the onset of genetic engineering in the 1980s allowed the possibility of rational design of bacteria to catabolize specific compounds, which could eventually be released into the environment as bioremediation agents. The complexity of this endeavour and the lack of fundamental knowledge nonetheless led to the virtual abandonment of such a recombinant DNA-based bioremediation only a decade later. In a twist of events, the last few years have witnessed the emergence of new systemic fields (including systems and synthetic biology, and metabolic engineering) that allow revisiting the same environmental pollution challenges through fresh and far more powerful approaches. The focus on contaminated sites and chemicals has been broadened by the phenomenal problems of anthropogenic emissions of greenhouse gases and the accumulation of plastic waste on a global scale. In this article, we analyze how contemporary systemic biology is helping to take the design of bioremediation agents back to the core of environmental biotechnology. We inspect a number of recent strategies for catabolic pathway construction and optimization and we bring them together by proposing an engineering workflow.
|
|
Scooped by
mhryu@live.com
April 12, 2018 1:09 PM
|
Our ability to generate bacterial strains with unique and increasingly complex functions has rapidly expanded in recent times. The capacity for DNA synthesis is increasing and costing less; new tools are being developed for fast, large-scale genetic manipulation; and more tested genetic parts are available for use, as is the knowledge of how to use them effectively. These advances promise to unlock an exciting array of 'smart' bacteria for clinical use but will also challenge scientists to better optimize preclinical testing regimes for early identification and validation of promising strains and strategies. Here, we review recent advances in the development and testing of engineered bacterial diagnostics and therapeutics. We highlight new technologies that will assist the development of more complex, robust and reliable engineered bacteria for future clinical applications, and we discuss approaches to more efficiently evaluate engineered strains throughout their preclinical development.
|
|
Scooped by
mhryu@live.com
April 11, 2018 12:37 AM
|
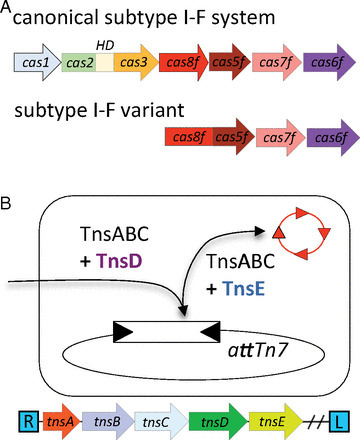
CRISPR-Cas is an adaptive immunity system that protects bacteria and archaea from mobile genetic elements. We present comparative genomic and phylogenetic analysis of minimal CRISPR-Cas variants associated with distinct families of transposable elements and develop the hypothesis that such repurposed defense systems contribute to the transposable element propagation by facilitating transposition into specific sites. Thus, these transposable elements are predicted to propagate via RNA-guided transposition, a mechanism that has not been previously described for DNA transposons.
|
|
Scooped by
mhryu@live.com
April 11, 2018 12:19 AM
|
The number of microbial genomes sequenced each year is expanding rapidly, in part due to genome-resolved metagenomic studies that routinely recover hundreds of draft-quality genomes. Rapid algorithms have been developed to comprehensively compare large genome sets, but they are not accurate with draft-quality genomes. Here we present dRep, a program that reduces the computational time for pairwise genome comparisons by sequentially applying a fast, inaccurate estimation of genome distance, and a slow, accurate measure of average nucleotide identity. dRep achieves a 28 × increase in speed with perfect recall and precision when benchmarked against previously developed algorithms. We demonstrate the use of dRep for genome recovery from time-series datasets. Each metagenome was assembled separately, and dRep was used to identify groups of essentially identical genomes and select the best genome from each replicate set. This resulted in recovery of significantly more and higher-quality genomes compared to the set recovered using co-assembly.
|
|
Scooped by
mhryu@live.com
April 10, 2018 11:52 PM
|
Dissimilatory perchlorate reduction is an anaerobic respiratory pathway that in communities might be influenced by metabolic interactions. Because the genes for perchlorate reduction are horizontally transferred, previous studies have been unable to identify uncultivated perchlorate-reducing populations. Here we recovered metagenome-assembled genomes from perchlorate-reducing sediment enrichments and employed a manual scaffolding approach to reconstruct gene clusters for perchlorate reduction found within mobile genetic elements. De novo assembly and binning of four enriched communities yielded 48 total draft genomes. In addition to canonical perchlorate reduction gene clusters and taxa, a new type of gene cluster with an alternative perchlorate reductase was identified. Phylogenetic analysis indicated past exchange between these gene clusters, and the presence of plasmids with either gene cluster shows that the potential for gene transfer via plasmid persisted throughout enrichment. However, a majority of genomes in each community lacked perchlorate reduction genes. Putative chlorate-reducing or sulfur-reducing populations were dominant in most communities, supporting the hypothesis that metabolic interactions might result from perchlorate reduction intermediates and byproducts. Other populations included a novel phylum-level lineage (Ca. Muirbacteria) and epibiotic prokaryotes with no known role in perchlorate reduction. These results reveal unexpected genetic diversity, suggest that perchlorate-reducing communities involve substantial metabolic interactions, and encourage expanded strategies to further understand the evolution and ecology of this metabolism.
|
|
Scooped by
mhryu@live.com
April 10, 2018 12:24 AM
|
Like all higher organisms, plants have evolved in the context of a microbial world, shaping both their evolution and their contemporary ecology. Interactions between plant roots and soil microorganisms are critical for plant fitness in natural environments. Given this co-evolution and the pivotal importance of plant–microbial interactions, it has been hypothesized, and a growing body of literature suggests, that plants may regulate the composition of their rhizosphere to promote the growth of microorganisms that improve plant fitness in a given ecosystem. Here, using a combination of comparative genomics and exometabolomics, we show that pre-programmed developmental processes in plants (Avena barbata) result in consistent patterns in the chemical composition of root exudates. This chemical succession in the rhizosphere interacts with microbial metabolite substrate preferences that are predictable from genome sequences. Specifically, we observed a preference by rhizosphere bacteria for consumption of aromatic organic acids exuded by plants (nicotinic, shikimic, salicylic, cinnamic and indole-3-acetic). The combination of these plant exudation traits and microbial substrate uptake traits interact to yield the patterns of microbial community assembly observed in the rhizosphere of an annual grass. This discovery provides a mechanistic underpinning for the process of rhizosphere microbial community assembly and provides an attractive direction for the manipulation of the rhizosphere microbiome for beneficial outcomes.
|