Your new post is loading...

|
Scooped by
mhryu@live.com
Today, 2:26 PM
|
During the last few years, the body of data on proteins is expanding almost exponentially with the development of advanced methods for gene sequencing, protein structure determination, particularly by cryoelectron microscopy, and structure prediction using artificial intelligence-based approaches. These developments create the potential for a comprehensive exploration of the protein universe, the entirety of the proteins existing in the biosphere. Elucidation of the relationships among proteins including the most distant ones, where only the core fold is shared, is crucial for understanding protein functions, folding mechanisms, and evolution, as well as the evolution of cellular life forms and viruses. In this brief review, we discuss methods that shaped the field of protein bioinformatics, first, through comparative sequence analysis, and the recent developments in protein structure prediction that transformed the state of the art in the comparative analysis of distantly related proteins. The combination of the rapidly growing databases of genome and metagenome sequences with sensitive methods for sequence comparison and the new generation of structure analysis tools can make charting the protein universe at the structural level a realistic goal.
|
|
Scooped by
mhryu@live.com
Today, 2:09 PM
|
Pseudomonas putida is a plant-beneficial rhizobacterium that encodes multiple type-VI secretion systems (T6SS) to outcompete phytopathogens in the rhizosphere. Among its antibacterial effectors, Tke5 (a member of the BTH_I2691 protein family) is a potent pore-forming toxin that disrupts ion homeostasis without causing considerable membrane damage. Tke5 harbors an N-terminal MIX domain, which is required for T6SS-dependent secretion in other systems. Many MIX domain-containing effectors require T6SS adaptor proteins (Tap) for secretion, but their molecular mechanisms of adaptor-effector binding remain elusive. Here, we report the 2.8 Å cryo-EM structure of the Tap3-Tke5 complex of P. putida strain KT2440, providing structural and functional insights into how effector Tke5 is recruited by its cognate adaptor protein Tap3. Functional dissection shows that the α-helical region of Tke5 is sufficient to kill intoxicated bacteria, while its β-rich region likely contributes to target membrane specificity. These findings delineate a mechanism of BTH_I2691 proteins for Tap recruitment and toxin activity, contributing to our understanding of a widespread yet understudied toxin family.
|
|
Scooped by
mhryu@live.com
Today, 1:31 AM
|
Synthetic rewriting technologies, encompassing large-scale DNA assembly, transfer, maintenance, and rearrangement, enabled de novo synthesis or large-scale modifications of genomes. While significant progress has been made in model organisms of viruses, bacteria, and unicellular eukaryotes, their development in mammalian cells faces unique challenges. This review summarizes key breakthroughs in synthetic rewriting technologies, including megabase (Mb)-scale assembly of human DNA, yeast-mediated transfer methods, bottom-up human artificial chromosomes (HACs), and genome-scale rearrangement, along with emerging applications in constructing models and decoding genomes for mammals. These tools will expand functional engineering in mammals and deepen mechanistic insights into complex biological systems. Synthetic DNA rewriting technologies enable de novo synthesis or large-scale modification of genomes. Here the authors discuss key advances in the DNA rewriting toolbox at the level of large-scale DNA assembly, transfer, maintenance, and rearrangement, whilst highlighting emerging applications.
|
|
Scooped by
mhryu@live.com
Today, 12:39 AM
|
Gene duplication has played a critical role in the evolutionary history of proteins, enabling complex multimers to emerge from simpler precursors. Yet in protein engineering, current methods for directed evolution do not exploit gene duplication, hampering access to the vast array of diverse variants that are only enriched in the presence of a wild-type copy. We establish a directed evolution strategy for multimeric proteins that harnesses gene duplication to compensate for metabolic burden and self-assembly fitness, allowing previously inaccessible variants to be enriched. Starting from a homomeric 240-mer capsid, gene duplication enables selection of both extreme homomeric variants and obligate heteromers. This strategy significantly expands engineering access to diverse high-performing variants, while also supporting a plausible model for evolutionary diversification of higher-order multimers in nature.
|
|
Scooped by
mhryu@live.com
Today, 12:34 AM
|
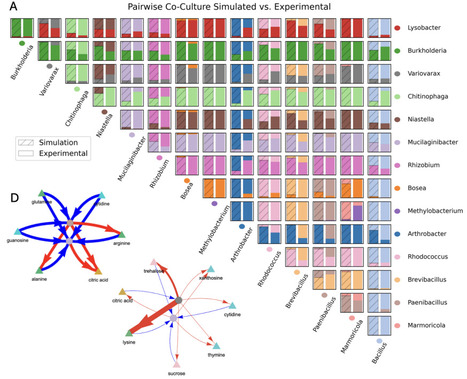
Synthetic microbial ecology aims at designing communities with desired properties based on mathematical models of individual organisms. It is unclear whether simplified models harbor enough detail to predict the composition of synthetic communities in metabolically complex environments. Here, we use longitudinal exometabolite data of monocultures for 15 rhizosphere bacteria to parametrize a consumer-resource model, which we use to predict pairwise co-cultures and higher order communities. The capacity to artificially "switch off" cross-feeding interactions in the model demonstrates their importance in ecosystem structure. Leave-one-out and leave-two-out experiments demonstrate that pairwise co-cultures do not necessarily capture inter-species interactions within larger communities and broadly highlight the nonlinearity of interactions. Finally, we demonstrate that our model can be used to identify new sub-communities of three strains with high likelihood of coexistence. Our results establish hybrid mechanistic and data-driven metabolic models as a promising and extendable framework for predicting and engineering microbial communities.
|
|
Scooped by
mhryu@live.com
Today, 12:20 AM
|
Genetically encoded biosensors enable the monitoring of metabolite dynamics in living organisms. We present CoBiSe, a computational biosensor design approach using Constraint Network Analysis to identify optimal insertion sites for reporter modules in molecular recognition elements (MREs). Applied to the iron-binding protein DtxR from Corynebacterium glutamicum, CoBiSe identified a flexible connective loop (residues 138–150) for inserting the reporter module, resulting in IronSenseR, a novel ratiometric biosensor for ferrous iron (Fe2+). IronSenseR demonstrates high specificity for Fe2+ with dissociation constants of 1.78 ± 0.03 (FeSO4) and 2.90 ± 0.12 μM (FeCl2), while showing no binding to Fe3+ and other divalent cations. In vivo assessment in Escherichia coli, Pseudomonas putida, and Corynebacterium glutamicum confirmed IronSenseR’s capability to detect changes in the intracellular iron pool. The creation of IronSenseR underlines that by reducing search space and eliminating labor-intensive screening, CoBiSe streamlines biosensor development and enables precise creation of next-generation biosensors for diverse metabolites.
|
|
Scooped by
mhryu@live.com
January 12, 11:56 PM
|
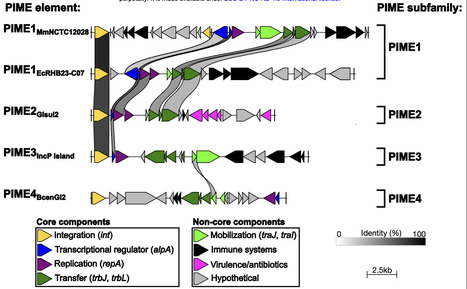
Integrative mobilizable elements (IMEs) are mobile genetic elements that reside stably integrated into chromosomes and rely on helper conjugative elements for horizontal transfer. Here, we identify and characterize a widespread family of IMEs, named Pseudomonadota Integrative Mobilizable Elements (PIMEs), which are distributed exclusively in the Pseudomonadota phylum. Genome and phylogenomic analyses reveal ∼1,000 putative PIMEs, comprising at least four distinct PIME subfamilies defined by distinctive genomic organizations and conserved hallmark features. Characterized PIMEs depend on helper conjugative plasmids of the incompatibility group P (IncP) and, upon induction, PIMEs excise, replicate and mobilize intra- and inter-species. Remarkably, the representative PIME and its helper conjugative plasmid engages in cross-complementation, revealing an unrecognized level of functional interplay between hijacker and helper element. We also demonstrate that PIMEs act as reservoirs of known and novel prokaryotic immune systems. Overall, our findings uncover an overlooked and disseminated family of IMEs, which likely plays an important role in bacterial ecology and evolution.
|
|
Scooped by
mhryu@live.com
January 12, 11:33 PM
|
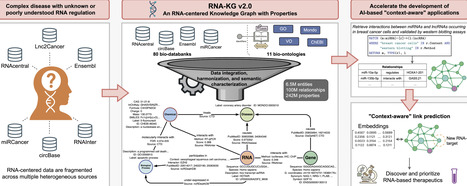
RNA-KG is a recently developed biomedical knowledge graph that integrates the interactions involving coding and non-coding RNA molecules extracted from public data sources. It can be used to support the classification of new molecules, identify new interactions through the use of link prediction methods, and reveal hidden patterns among the represented entities. In this paper, we propose RNA-KG v2.0, a new release of RNA-KG that integrates around 100M manually curated interactions sourced from 91 linked open data repositories and ontologies. Relationships are characterized by standardized properties that capture the specific context (e.g. cell line, tissue, pathological state) in which they have been identified. In addition, the nodes are enriched with detailed attributes, such as descriptions, synonyms, and molecular sequences sourced from platforms such as OBO ontologies, NCBI repositories, RNAcentral, and Ensembl. The enhanced repository enables the expression of advanced queries that take into account the context in which the experiments were conducted. It also supports downstream applications in RNA research, including ‘context-aware’ link prediction techniques that combine both topological and semantic information. Finally, the recent integration of RNA-KG relationships into the RNAcentral portal provides a powerful resource for linking RNA-centric relationships with non-coding gene expression in human tissues, RNA secondary structures, and their functional roles in biological pathways, which can accelerate the discovery of novel therapeutic targets.
|
|
Scooped by
mhryu@live.com
January 12, 11:19 PM
|
Early-life microbial exposures shape immune development and allergy risk. Food allergen sensitization, reflected by the presence of food allergen-specific immunoglobulin E (IgE), is an early indication of impaired immune tolerance. Here we show that early-life transmission of aromatic lactate-producing bifidobacteria strains in 147 children followed from birth to 5 years of age, facilitated by vaginal delivery, exposure to older siblings and exclusive breastfeeding for the first 2 months, led to increased levels of aromatic lactates in the infant gut. This microbiota–metabolite signature was inversely associated with the development of food allergen-specific IgE until 5 years and atopic dermatitis at 2 years. The observed effect was mediated by 4-hydroxy-phenyllactate, which inhibited IgE, but not IgG, production in ex vivo human immune cell cultures. Together, these findings define an early-life microbiota–metabolite–immune axis linking microbial transmission and feeding practices with reduced allergic sensitization. Vaginal birth, exclusive breastfeeding and early contact with siblings promote colonization of the infant gut with bifidobacteria capable of producing aromatic lactates, a microbial and metabolite signal that is inversely related to the risk of allergen-specific sensitization and dermatitis later in life.
|
|
Scooped by
mhryu@live.com
January 12, 11:03 PM
|
This study presents Bacillus subtilis T7 as the first known strain of B. subtilis capable of simultaneous lignin depolymerization and direct hydrogen production—a dual metabolic capability not previously reported in this species. B. subtilis T7 demonstrated 63.38% lignin degradation and 56.53% Azure B decolorization over seven days, with HPLC detection of aromatic intermediates—ferulic acid (0.85 mg/L) and vanillin (0.666 mg/L)—confirming active lignin catabolism. Agar-based assays revealed robust hydrolytic enzyme activities, including proteases (23.3 mm), cellulases (24.5 mm), amylases (14.2 mm), and xylanases (11.6 mm), surpassing those of many reported Bacillus strains. Whole-genome analysis confirmed a cascade of carbohydrate-active enzymes (CAZymes) including AA10 (lytic polysaccharide monooxygenases), AA3 (oxidoreductases), AA6 (quinone reductases), and CE1 (acetyl xylan esterases). These enzymes are associated with enhanced cellulolytic and xylanolytic activities, as well as increased lignin degradation. Batch fermentation experiments demonstrated that B. subtilis T7 produced hydrogen yields ranging from 0.53 to 1.41 mol H₂/mol substrate across various feedstocks, including xylose, glucose, carboxymethyl cellulose (CMC), starch, and untreated food waste. Xylose exhibited the highest volumetric productivity, achieving 274 mL H₂/g volatile solids, along with the most rapid production kinetics, indicating efficient metabolic utilization of this pentose sugar. In contrast, untreated food waste yielded the maximum molar hydrogen output of 1.41 mol H₂/mol substrate, attributable to its heterogeneous carbohydrate composition and lower average molecular weight, which likely enhanced enzymatic accessibility and substrate solubilization. These findings indicate that B. subtilis T7 encodes a functional [FeFe]-hydrogenase operon, along with an expanded repertoire of oxidative CAZymes, enabling it to bioprocess waste biomass into hydrogen without the need for syntrophic partners.
|
|
Scooped by
mhryu@live.com
January 12, 10:01 PM
|
Rhizosphere microbes benefit plant growth and health. How plant-microbe interactions regulate fruit quality remains poorly understood. Here, we elucidate the multi-level modulation of vitamin accumulation in tomato by flavonoid-mediated crosstalk between host plants and rhizosphere microbes. SlMYB12-overexpressing plants with up-regulated flavonoid biosynthesis accumulate higher levels of vitamins C and B6 in fruits compared to wild-type plants grown in natural soil. Flavonoid-mediated improvement of fruit quality depends on the presence of soil microbiomes and relates to rhizosphere enrichment of key taxa (e.g. Lysobacter). Multi-omics analyses reveal that flavonoids attract Lysobacter soli by stimulating its twitching motility and spermidine biosynthesis, which in turn boosts vitamin accumulation in fruits across tomato cultivars and soil types. RpoN acts as a dual regulator in L. soli that is responsive to flavonoids, controlling bacterial motility and spermidine production. Our study provides insight into flavonoid-mediated rhizosphere signalling and underscores plant-microbiome orchestration for improved tomato fruit quality. How fruit quality is regulated by plant microbiome remains poorly understood. Here, the authors reveal that flavonoids secreted by tomato roots can recruit specific soil microbes to the rhizosphere and stimulate spermidine biosynthesis, which can induce vitamin accumulation in tomato fruits.
|
|
Scooped by
mhryu@live.com
January 12, 9:45 PM
|
Heteroresistance is a transient resistance phenotype characterized by the presence of small subpopulations of bacterial cells with elevated antibiotic resistance within a susceptible main population. In gram-negative pathogens, heteroresistance is frequently caused by tandem amplification of genes encoding resistance proteins with low activity toward the antibiotic, a process commonly mediated by homologous recombination between flanking repeated sequences. However, the specific roles of individual recombination proteins in this mechanism remain largely undefined. In this study, we systematically evaluated the contribution of 19 recombination-associated genes to tandem amplification-driven heteroresistance in Escherichia coli. A clinical plasmid causing tobramycin heteroresistance by tandem amplification of the aac(3)-IId gene was conjugated into recombination gene-deficient mutants and the wild-type parental strain. While heteroresistance was observed with all mutants, the frequency of resistant subpopulations was decreased in recA and recB mutants, and a shift in resistance mechanism toward increased plasmid copy number and resistance mutations was observed. Partially reduced frequencies of tandem amplifications and a shift toward other heteroresistance mechanisms were also observed with recC, recJ, ruvA, and ruvC mutants, whereas other deletions of recombination genes had no or little impact on tandem amplifications. These findings identify RecABC as a key pathway in heteroresistance via tandem amplification, but even when these genes are deleted, resistant subpopulations can still be generated by other mechanisms.
|
|
Scooped by
mhryu@live.com
January 12, 3:53 PM
|
Bacterial microcompartments (BMCs) are pseudo-organelles that sequester metabolic enzymes, intermediates, and/or gases within the bacterial cytosol. One model BMC is the carboxysome (CB). CBs facilitate rubisco-driven fixation of CO2, increasing efficiency and maximizing the phosphoglycerate output in CB-containing bacteria. The α-CBs are of particular interest due to their small size and relative simplicity, making them ideal targets for bioengineering applications. These CBs were the first BMC observed and have been a long-studied model; however, they are challenging to study in native systems and in purified samples. Recent advances in cryogenic electron microscopy and cryogenic electron tomography have resulted in many new published structures of the shell proteins, shell assemblies, and cargo organization within the CB. These new insights have advanced the field’s understanding of important structural interfaces, shed insights into once unknown domain functions, and the complex mechanisms involved in assembly and maintenance of the CB. This review highlights recently published structures of α-CB proteins and the functional and mechanistic findings of these studies.
|
|
|
Scooped by
mhryu@live.com
Today, 2:22 PM
|
Translation is a fundamental process for every living organism. In plants, the rate of translation is tightly modulated during development and in responses to environmental cues. However, it is challenging to measure the actual translation state of the tissues in vivo. Here, we report the introduction of an in vivo translation marker based on bimolecular fluorescence complementation, the Ribo-BiFC. We combined a method originally developed for the fruitflies with an improved low background split-mVenus BiFC system previously described in plants. We labelled small subunit ribosomal proteins (RPS) and large subunit ribosomal proteins (RPL) of Arabidopsis thaliana with fragments of the mVenus fluorescent protein (FP). We tested the Ribo-BiFC method using transiently expressed recombinant ribosomal proteins in epidermal cells of Nicotiana benthamiana. The BiFC-tagged ribosomal proteins complemented the mVenus molecule and were detected by fluorescence microscopy, potentially visualizing the close proximity of translating assembled 80S ribosomal subunits. Although the resulting signal is less intense than that of known interactors, its detection points to the functionality of the system. This Ribo-BiFC approach has further potential for use in stable transgenic lines in enabling the visualization of translational rate in plant tissues and changing translation dynamics during plant development, under abiotic stress or in different genetic backgrounds.
|
|
Scooped by
mhryu@live.com
Today, 1:12 PM
|
Nutrient crossfeeding critically governs microbiome–host interactions and ecosystem stability. Cobamides, synthesized only by prokaryotes, offer a powerful and tractable model for studying nutrient-mediated interdependencies in soil food webs; however, their ecological role in sustaining soil health remains unclear. Here, we construct the Soil Cobamide Producer database (SCP v.1.0) by integrating over 48,000 metagenomic and genomic datasets from 1,123 sampling sites. This database catalogs phylogenetically diverse prokaryotes (19 phyla, 302 genera) with cobamide biosynthetic potential. Using this resource, we identify host-specific colonization patterns of cobamide-producing microbes in fauna. These microbes also carry diverse functional traits that may contribute to trophic cascades and microbial community stability. In an Enchytraeid model, these colonizers support host development, modulate gene expression, and promote gut stability through transkingdom interactions, with cobamide biosynthesis serving as one representative trait among multiple microbial functions. At macroecological scales, cobamide-producing microbes occur across relatively high trophic levels, reflecting a broader principle of nutrient transfer that may also apply to other essential metabolites. This framework provides a general basis for studying nutritional microbes in soil food webs and advances One Health research. Soil life relies on microbial nutrient exchange to maintain ecosystem balance. This study shows that cobamide-producing microbes act as essential connectors in soil food webs, supporting host health and highlighting cross-kingdom links central to One Health.
|
|
Scooped by
mhryu@live.com
Today, 1:27 AM
|
High-throughput sequencing and computational protein design have created a growing gap between the discovery of new proteins and their functional characterization. In many instances, functional characterization requires one-to-one measurements—such as when detailed biochemical insights are desired or pooled selections are not possible—necessitating that individual variants be isolated and assayed. A major barrier to closing this gap is the cost to directly synthesize individual genes, which remains prohibitively expensive ($10–100 per sequence) and restricts these studies to small subsets of relevant variants, leaving many sequences without functional annotation. To address this, we developed user-defined Sorted Mutants (uSort-M), which combines pooled DNA synthesis, automated cell sorting of transformed Escherichia coli, and long-read sequencing to rapidly isolate and identify variants from diverse libraries. uSort-M can isolate, sequence, and validate individual variants from pooled libraries produced via diverse existing methods including multiplex assembly, error-prone PCR, or pooled QuickChange mutagenesis. Sorting single bacterial clones into 384-well plates is efficient: eight plates (3,072 wells) can be filled in 1–2 hours, with up to 90% of wells yielding monoclonal cultures. Commercial long-read sequencing enables accessible, fast, and cost-effective identification of individual sequences from isolated clones while tolerating wide variation in fragment length and diversity across the library. Applying this workflow to a 328-member scanning mutagenesis library of a 300-bp gene recovered 96% of desired variants at fivefold lower cost than traditional synthesis. Numerical simulations identify key parameters governing library recovery and enable accurate prediction of the sampling effort required to achieve target coverage. As library size increases, this workflow offers substantial savings over traditional gene synthesis or cloning. Due to its generalizability, efficiency, and reliance on standard instrumentation, uSort-M removes a key barrier to large-scale protein functional characterization.
|
|
Scooped by
mhryu@live.com
Today, 12:36 AM
|
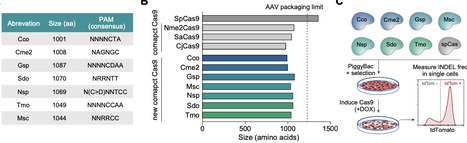
The development of compact and efficient CRISPR-Cas systems is crucial for biomedical and therapeutic genome editing, particularly in vivo applications based on viral delivery. Here, we performed a comparative functional screen of seven Cas9 orthologs to systematically evaluate their genome editing activity in mammalian cells. Among these, Cme2, a 1008 amino acid nuclease recognizing a 5' NAGNGC PAM, emerged as a promising candidate based on its compact size and baseline editing activity. To overcome its limited native efficiency, we employed a dual engineering approach combining sgRNA scaffold optimization and rational protein mutagenesis. The resulting variant, enCme2, exhibits markedly improved editing efficiency across multiple loci in both mouse and human cells while maintaining extremely high specificity and minimal off-target activity. Importantly, the small size of enCme2 permits packaging of the complete system into a single rAAV vector, enabling efficient genome editing/HDR in in vivo tissues and mouse embryos, and facile generation of transgenic models. These results establish enCme2 as a compact, precise, and AAV-compatible genome editing platform with broad applicability for in vivo research and therapeutic approaches, especially where high specificity is desirable.
|
|
Scooped by
mhryu@live.com
Today, 12:29 AM
|
The rapid advancement of environmental sequencing technologies, such as metagenomics, has significantly enhanced our ability to study microbial communities. The eubiotic composition of these communities is crucial for maintaining ecological functions and host health. Species diversity is only one facet of a healthy community’s organization; together with abundance distributions and interaction structures, it shapes reproducible macroecological states, that is, joint statistical fingerprints that summarize whole-community behavior. Despite recent developments, a theoretical framework connecting empirical data with ecosystem modeling is still in its infancy, particularly in the context of disordered systems. Here, we present a novel framework that couples statistical physics tools for disordered systems with metagenomic data, explicitly linking diversity, interactions, and stability to define and compare these macroecological states. By employing the generalized Lotka–Volterra model with random interactions, we reveal two different emergent patterns of species interaction networks and species abundance distributions for healthy and diseased microbiomes. On the one hand, healthy microbiomes have similar community structures across individuals, characterized by strong species interactions and abundance diversity consistent with neutral stochastic fluctuations. On the other hand, diseased microbiomes show greater variability driven by deterministic factors, thus resulting in less ecologically stable and more divergent communities. Our findings suggest the potential of disordered system theory to characterize microbiomes and to capture the role of ecological interactions on stability and functioning.
|
|
Scooped by
mhryu@live.com
Today, 12:14 AM
|
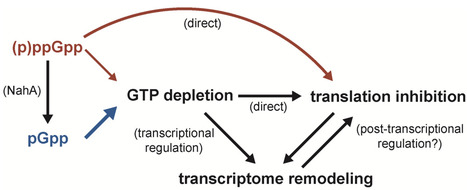
Bacteria produce the alarmone nucleotides (p)ppGpp during stress to affect replication, transcription, translation, and metabolism. Recently, pGpp was identified as a third alarmone that is produced from the hydrolysis of (p)ppGpp. Although pGpp is a major component of bacterial stress responses, its precise role in mediating these responses is poorly understood. ppGpp and pppGpp bind translation GTPases and therefore directly affect translation to conserve resources during periods of stress. Here, we show that while pGpp is a weaker inhibitor of protein synthesis than ppGpp and pppGpp in vitro, pGpp production in the model Gram-positive bacterium Bacillus subtilis leads to faster translation inhibition in vivo. Faster translation inhibition is accompanied by greater levels of disengaged ribosomal subunits and hibernating ribosome dimers, suggesting that translation initiation is strongly inhibited. We show that alarmone production in vivo causes a severe depletion of GTP, which is sufficient for translation inhibition. Finally, we find that pGpp production also causes more robust transcriptome remodeling than (p)ppGpp production. This work supports a model that implicates all three alarmones in translation inhibition via GTP depletion.
|
|
Scooped by
mhryu@live.com
January 12, 11:42 PM
|
Soil bacteria are critical to agricultural productivity and play a central role in several biogeochemical cycles. These organisms frequently experience desiccation, which deprives them of access to both water and nutrients. Desiccation eliminates the aqueous connections between soil pores, removing pathways for nutrient diffusion. Well-studied yet resource intensive water stress responses like osmolyte synthesis may thus be impractical in unsaturated environments. Accordingly, we observed how the rhizobacterium Pseudomonas synxantha 2-79 responds to co-occurring water and nutrient limitation at the single-cell level to understand what role osmolyte synthesis may play in its desiccation response. We constructed a transcriptional reporter to track P. synxanthas osmolyte response and collected extensive morphological and reporter expression data through experiments designed to mimic different rates and extents of soil drying. Only actively growing cells responded to an osmotic shock by synthesizing osmolytes: this response was not observed when we pre-starved bacteria. Soluble nutrient diffusivity in soil is restricted even when there is sufficient water to keep bacterial cells hydrated, so this pre-starved condition reflects gradual drying in which starvation precedes water stress. Despite the lack of osmolyte synthesis, prior starvation enhanced P. synxanthas ability to recover from osmotic stress once water and nutrients were restored. These results suggest that cellular changes associated with the response to starvation that go beyond osmolyte synthesis play an important role in microbial desiccation tolerance.
|
|
Scooped by
mhryu@live.com
January 12, 11:31 PM
|
In vitro antibiotic testing is important for guiding therapy and drug development. Current methods are focused on growth inhibition in bulk bacterial populations but often fail to accurately predict treatment responses. Here we introduce Antimicrobial Single-Cell Testing (ASCT), a large-scale live-cell imaging approach that quantifies bacterial killing in real time at single-cell resolution. By tracking over 140 million mycobacteria and analysing ~20,000 time–kill curves, we identify key determinants of antibiotic killing and its clinical relevance. For Mycobacterium tuberculosis, we found that drug-specific killing dynamics in starved bacteria, rather than growth inhibition or killing of growing cells, predict regimen efficacy in mice and humans. Extending this approach to Mycobacterium abscessus and comparing 405 bacterial strains, we show that antibiotic killing is also a genetically encoded bacterial trait (drug tolerance). We demonstrate that tolerance patterns cluster by antibiotic targets, identify a phage protein that modulates antibiotic killing, and show that strain-specific killing dynamics are associated with individual patient outcomes independent of drug resistance. Together, these findings establish a framework that reveals how drug properties and bacterial diversity shape treatment responses, offering a path to more effective and personalized therapies. Via high-throughput imaging and tracking over 140 million single mycobacteria, the authors show that drug- and strain-specific killing predict treatment outcomes, with potential to improve drug development and personalized therapy.
|
|
Scooped by
mhryu@live.com
January 12, 11:13 PM
|
Malonyl-CoA serves as a central precursor for the biosynthesis of diverse high-value compounds, including lipids, organic acids, and polyketides, in engineered microbial fermentation systems. However, insufficient malonyl-CoA supply often limits the production of these products. Intracellular malonyl-CoA levels are tightly regulated by the activities of acetyl-CoA carboxylase (ACC) and fatty acid synthesis pathway enzymes. Although strategies have been developed to redirect malonyl-CoA flux from fatty acid biosynthesis toward desired products, native ACC-mediated synthesis remains constrained by slow kinetics, complex regulation, and ATP consumption. To overcome these limitations, two alternative malonyl-CoA biosynthetic pathways have recently been developed. The malonate assimilation pathway enables direct uptake and CoA ligation of exogenous malonate, providing precise control over malonyl-CoA metabolism. The non-carboxylative malonyl-CoA (NCM) pathway converts pyruvate to malonyl-CoA through a novel intermediate, eliminating both ATP and CO2 loss while simultaneously regenerating NADPH. This review highlights recent advances in these two alternative malonyl-CoA biosynthetic pathways and their applications across diverse microbial hosts, underscoring their potential to enhance the sustainable production of valuable biochemicals.
|
|
Scooped by
mhryu@live.com
January 12, 10:18 PM
|
Hyperlysinemia is a life-threatening metabolic disorder that requires the continuous clearance of lysine. Engineered probiotics capable of degrading lysine in the gut represent a promising therapeutic strategy. However, the introduction of heterologous metabolic pathways can impose a substantial fitness burden on the bacterial host, potentially compromising the therapeutic efficacy. Current screening methods fail to adequately assess this pathway-induced stress. Therefore, optimizing methods to evaluate bacterial fitness after pathway modification is essential for developing effective bacterial therapies. Here, we present a label-free phenotypic screening approach using Fourier transform infrared (FTIR) spectroscopy to evaluate the physiological burden imposed by two distinct lysine catabolism pathways engineered E. coli Nissle 1917 (EcN): the plant-derived bifunctional enzyme LKR-SDR and the yeast-derived two-enzyme cascade Lys2-Lys5. Employing FTIR under lysine stress mimicking pathological concentrations, decoded pathway-specific stress signatures, and molecular resilience. Probiotics expressing LKR-SDR exhibited severe multisystem damage, including proteotoxicity, lipid peroxidation, and significant nucleic acid stress. In contrast, the Lys2-Lys5 strain demonstrated superior resilience, maintained structural integrity, and exhibited adaptive metabolic changes, primarily through lipid membrane remodeling. This study establishes FTIR spectroscopy as a rapid screening platform that identifies the Lys2-Lys5 pathway as optimal for probiotic therapies. By directly linking spectroscopic signatures to cellular fitness, FTIR spectroscopy accelerates the rational development of durable microbial therapeutics for inborn metabolic disorders.
|
|
Scooped by
mhryu@live.com
January 12, 9:48 PM
|
Streptomyces produce a multitude of secondary metabolites, which have been exploited in drug discovery campaigns for more than three-quarters of a century. Our understanding of microbial physiology has been revolutionized by genome sequencing and large-scale functional studies. Technology for genome-wide investigations in Streptomyces species, however, has lagged behind that for other bacterial systems, hindering exploitation of unprecedented quantities of genomic data. Here, we develop a platform for en masse CRISPRi-seq for Streptomyces spp. By performing CRISPRi-seq with 2,160 unique sgRNAs targeting all operons (432 operons) encoding membrane transporters (629 genes) representing 1.1Mb of the 6.8Mb genome for S. albidoflavus, combined with hit validation, we discovered that only a small proportion (13 of 432 operons, 25 kb) contribute positively to fitness. Our work provides both a first-in-class platform for high-throughput functional genomics and a generalized blueprint for en masse screens in Streptomyces species.
|
|
Scooped by
mhryu@live.com
January 12, 4:05 PM
|
Ammonia present in the environment is a major source of nitrogen, but it can be toxic to bacteria. While the biochemical mechanisms involved in the metabolic detoxification of cellular ammonia are well understood, little is known about how bacteria manage toxic external ammonia to survive, especially when ammonia is present as a waste product at high concentrations. Here, we demonstrate that a two-component system consisting of the sensor kinase GrtK and the response regulator GrtR is responsible for sensing and neutralizing toxic environmental ammonia produced as a waste product by the rice pathogen Burkholderia glumae. The growth of null mutants of grtK or grtR was inhibited in amino acid-rich media such as Luria-Bertani medium, but no growth inhibition was observed in amino acid-free media. The expression of obcAB, responsible for the biosynthesis of the previously known neutralizing agent oxalate, was dependent upon external ammonia concentration in a GrtR-dependent manner. Significant changes in fluorescence were observed when cells of B. glumae carrying a recombinant plasmid of the modified circular permutation GFP gene fused to grtK were incubated with compounds containing ammonium, suggesting that GrtK interacts selectively with external ammonia. Transcriptome analysis of grtK and grtR mutants also showed that GrtK and GrtR are involved in the metabolic detoxification of cellular ammonia as well. These results indicate that GrtK is an external ammonia sensor that is not a member of the ammonia transporter protein family and works together with the response regulator GrtR to counter the risk posed by its own metabolism.
|