Your new post is loading...

|
Scooped by
mhryu@live.com
Today, 1:41 AM
|
Spray-induced gene silencing (SIGS) provides a sustainable, highly targeted alternative to chemical pesticides. This review summarizes recent advances in dsRNA technology through a design-to-delivery framework. Key focuses include bioinformatics-driven multi-target dsRNA design for improved stability and efficacy, cost-effective microbial and cell-free synthesis platforms for scalable production, and nanocarriers that protect dsRNA from degradation while enhancing delivery. Integrating rational design, efficient production, and precision nanodelivery will accelerate SIGS from laboratory concept to practical, eco-friendly tool for global crop protection and sustainable agriculture. A review summarizes advances in spray-induced gene silencing (SIGS), covering rational dsRNA design for efficacy, scalable microbial/cell-free production, and nanodelivery systems for stable, effective crop protection.
|
|
Scooped by
mhryu@live.com
Today, 1:34 AM
|
Biological N2O production from organic nitrogen is generally assumed to require canonical nitrification, which generates oxidized nitrogen that subsequently fuel denitrification. Whether this paradigm universally applies to nitrogen-rich microbial communities remains unclear. Here, we investigated N2O production across an industrial poultry manure composting process and found that substantial N2O formation occurred despite the apparent absence of canonical ammonia oxidation. Neither allylthiourea inhibition nor metagenomic analyses provided evidence for ammonia-oxidizing microorganisms or their activity. Instead, metagenomic analyses identified abundant bacterial nitric oxide synthase (bNos) genes, many of which were phylogenetically affiliated with Bacilli, the dominant bacterial group throughout composting. Physiological experiments with Bacillus isolates demonstrated a nitrification-independent route in which L-arginine was oxidized to NO2⁻/NO3⁻, consistent with bNOS-mediated NO formation followed by abiotic oxidation. Recovery of 15N-labelled N2O following 15NO2⁻ addition established NO2⁻ as an immediate precursor of aerobically produced N2O, confirming that the oxidized nitrogen generated through this alternative route subsequently fueled denitrification. Metagenomic analyses further revealed extensive denitrification potential but comparatively low nosZ abundance. Together, these findings identify a previously overlooked route linking organic nitrogen turnover to denitrification independently of canonical nitrification, thereby expanding current models of microbial N2O production in composts and potentially other protein-rich thermophilic environments.
|
|
Scooped by
mhryu@live.com
Today, 12:43 AM
|
Cell division is a fundamental process essential for life, underpinning reproduction, development and tissue maintenance across all organisms and enabling population growth and evolutionary adaptation. Recreating this capability is, therefore, a central challenge in bottom–up synthetic biology, wherein the aim is to construct functional synthetic cells. In recent years, substantial progress has been made toward building a synthetic divisome through partial reconstitution of the protein machinery underlying cell division in vitro. Here, we review current strategies to mimic the key stages of division: symmetry breaking to define the division site, membrane deformation to drive constriction and, thus, shape changes of the cell, and the final abscission event. We critically assess the successes and limitations of these approaches and discuss how integrating multiple modules may enable the realization of a minimal, functional division system for synthetic cells. Cell division consists of three elementary steps: symmetry breaking, membrane constriction and abscission. Here, we review current strategies for achieving these steps in synthetic cells, assessing their successes and limitations with a view to integrating them within a divisome for synthetic cells.
|
|
Scooped by
mhryu@live.com
Today, 12:04 AM
|
Root system architecture and root hairs highly influence plant resource uptake, yet their simultaneous quantification at the whole-plant scale remains challenging due to the conflicting requirements of high-resolution imaging and non-destructive, repeated measurements. Here, we present a plant growth and imaging system based on A3 sized flatbed rhizotrons, combined with a non-machine-learning image analysis workflow implemented in R, that enables repeated, in situ quantification of barley root system architecture and accompanied visible projected root hair area (VP root hair area) over three weeks of growth. The system produces integrated outputs highlighting both whole-root architecture and the spatial distribution of surrounding root hairs from single images. Validation of the image analysis algorithm showed good segmentation performance, with average Matthews Correlation Coefficient values of 0.68 for projected root area and 0.65 for VP root hair area. To demonstrate its experimental applicability, the system was used to assess root growth and root hair responses under controlled environmental conditions, combining three irrigation regimes (2, 4, and 6 irrigation events per day) with three dry bulk density levels (1.4, 1.5, and 1.6 g cm−3). In addition to whole-system metrics, the approach enables analysis of root hair expansion at individual root tips. This methodology provides a training-free method for integrated analysis of root traits e.g. projected root size, root growth, root diameter and root hairs under controlled physical conditions similar to soil, facilitating studies of root–soil interactions that require both spatial resolution and temporal continuity.
|
|
Scooped by
mhryu@live.com
July 13, 11:13 PM
|
Transposons are fundamental genetic elements that have profoundly shaped the architecture of eukaryotic genomes. Yeasts and filamentous fungi have emerged as important chassis organisms for bioingredient production in synthetic biology and metabolic engineering. In this review, we summarise the current understanding and future opportunities in the development of transposon-based tools for genome engineering in these fungal systems. Fungal inverted terminal repeat (ITR) DNA transposons, as well as long terminal repeat (LTR) and non-LTR retrotransposons, can accelerate genomic mutagenesis, facilitating the screening of superior genotypes and phenotypes. CRISPR-associated transposons (CASTs) hold considerable potential for site-specific integration of large transgenes, bypassing the limitations imposed by low homologous recombination (HR) efficiency in non-Saccharomyces hosts. Overall, transposon-based tools represent a valuable and underexplored avenue to accelerate genome engineering and strain development in yeasts and filamentous fungi.
|
|
Scooped by
mhryu@live.com
July 13, 11:01 PM
|
Protein language models, as well as models that incorporate structural information or homologous sequences, estimate the sequence likelihood p(sequence), which reflects the protein fitness landscape and is commonly used in mutation effect prediction and protein design. It is widely believed in deep learning field that larger models perform better across tasks. However, for fitness prediction, language model performance declines beyond a certain size, raising concerns about their scalability. Here we showed that model size, training dataset and stochastic elements can bias the predicted p(sequence) away from real fitness. Model performance on fitness prediction depends on how well p(sequence) matches evolutionary patterns in homologs, which is best achieved at a moderate p(sequence) level for most proteins. At extreme predicted wild-type sequence likelihoods, models predict uniformly low or high likelihoods for nearly all mutations, failing to reflect the real fitness landscape. Notably, larger models tend to predict proteins with higher p(sequence), which may exceed the moderate range and thus reduce performance. Our findings clarify the scaling behavior of protein models on fitness prediction and provide practical guidelines for their application and future development. Larger language models do not always perform better at fitness prediction. Hou et al. found that performance depends on whether model outputs match evolutionary patterns in homologs, which is best achieved at moderate predicted sequence likelihoods.
|
|
Scooped by
mhryu@live.com
July 13, 10:37 PM
|
Cell-free protein synthesis (CFPS) offers rapid protein production, yet yield variability from volume constraints and batch-to-batch differences remains a challenge. Bayesian optimization (BO) identifies optimal points under fixed constraints but does not address constraint variability. Here we use Gaussian process (GP) predictive uncertainty in two complementary directions, lower and upper confidence bounds for exploitation and exploration, to design condition spaces under variable constraints. Closed-loop optimization of two CFPS systems improved green fluorescent protein (GFP) yield beyond the standard condition within a few rounds. For exploitation, we defined a yield assurance space (YAS), a low-uncertainty set of conditions selected to exceed a conservative yield threshold. For exploration, we proposed Pareto front expansion under tightened volume constraints via uncertainty-driven sampling. This approach illustrates how GP predictive uncertainty can support both assured condition design and targeted exploration under altered constraints.
|
|
Scooped by
mhryu@live.com
July 13, 10:13 PM
|
Identifying bacterial antimicrobial resistance (AMR) is critical for diagnostics and treatment, but resistance is a complex trait arising from myriad mechanisms spanning multiple molecular scales. Existing computational approaches often function as black boxes and rarely explore cross-species or multi-drug patterns. We developed amR, an integrated R package suite that provides a complete framework from bacterial genome data curation to interpretable AMR predictions, enabling identification of resistance mechanisms across species and drugs. The amR R package suite contains three modular packages. amRdata downloads genomes and paired antimicrobial susceptibility testing data from BV-BRC and processes them, constructs pangenomes, and extracts features at gene/protein cluster, protein domain, annotated Clusters of Orthologous Groups and ResFinder AMR-associated features, and structural variant scales; data are stored in memory-efficient formats (Parquet, DuckDB). amRml trains interpretable machine learning models per species-drug combination, calculates feature importance and performance metrics, and provides rich ground for hypothesis generation and mechanism discovery. amRviz provides an interactive Shiny dashboard to explore metadata distributions and model performance across species and drugs, visualize top predictive AMR features, and analyze cross-model patterns across geographic/temporal strata. We apply the suite to Shigella sonnei, achieving a median Matthews Correlation Coefficient of 0.89 across 23 drugs and drug classes. With thousands of genomes, multi-scale features, and interpretable models, amR provides an accessible, comprehensive framework for AMR research. The amR package suite is installable via GitHub (https://github.com/JRaviLab/amR; BSD-3-Clause license).
|
|
Scooped by
mhryu@live.com
July 13, 5:29 PM
|
Understanding the mechanisms and structural mappings between molecules and pathway classes are critical for design of reaction predictors for synthesizing new molecules. This article studies the problem of prediction of classes of metabolic pathways (series of chemical reactions occurring within a cell) in which a given biochemical compound participates. We apply a hybrid machine learning approach consisting of graph convolutional networks used to extract molecular shape features as input to a random forest classifier. In contrast to previously applied machine learning methods for this problem, our framework automatically extracts relevant shape features directly from input SMILES representations, which are atom-bond specifications of chemical structures composing the molecules. Our method is capable of correctly predicting the respective metabolic pathway class of 95.16% of tested compounds, whereas competing methods only achieve an accuracy of 84.92% or less. Furthermore, our framework extends to the task of classification of compounds having mixed membership in multiple pathway classes. Our prediction accuracy for this multi-label task is 95.62%. We analyze the relative importance of various global physicochemical features to the pathway class prediction problem and show that simple linear/logistic regression models can predict the values of these global features from the shape features extracted using our framework.
|
|
Scooped by
mhryu@live.com
July 13, 2:45 PM
|
Antimicrobial resistance is an escalating global threat, with soils serving as reservoirs and conduits for the dissemination of antibiotic resistance genes (ARGs). Pesticide use in agriculture contributes to ARG proliferation, and ~60% of agricultural soils contain multiple pesticide residues. However, how pesticide diversity influences ARG dynamics in active microbial populations (active ARGs) remains unclear. Here, we evaluate the effects of pesticide diversity on active soil ARGs through a long-term field experiment integrating bioorthogonal non-canonical amino acid tagging (BONCAT), fluorescence-activated cell sorting (FACS), and metagenomics. We show that both low and high pesticide diversity significantly increase active ARG abundance relative to untreated control, whereas total ARG levels remain largely unchanged. The underlying mechanisms differ with pesticide diversity. At low diversity, active ARG co-selection via efflux pumps in Acinetobacter baumannii is a prominent mechanism. At high diversity, elevated reactive oxygen species and SOS responses promote horizontal gene transfer of active ARGs, as validated by culture experiments. These findings demonstrate that increasing pesticide diversity accelerates the emergence and dissemination of active ARGs, highlighting the need for integrated pesticide management strategies that consider both application intensity and diversity to mitigate resistance risks under the One Health framework. Here, the authors conduct a long-term field experiment to show that applying diverse pesticide mixtures increases active antibiotic resistance genes in soils. Low pesticide diversity co-selects for specific resistance genes, whereas high diversity accelerates spread between bacteria via horizontal gene transfer.
|
|
Scooped by
mhryu@live.com
July 12, 1:40 PM
|
Microbial defense systems are central to virus-host interactions and thus to microbial survival, but remain poorly studied in microbial communities of extreme environments. Here we examine the repertoire and distribution of microbial defense genes in hydrothermally influenced deep subsurface sediments and rocks of the Guaymas Basin (Gulf of California). Restriction–modification (RM) and abortive infection (Abi) systems were broadly distributed across the examined sediment depths, and CRISPR–Cas systems were primarily detected within temperate surficial sediments. Prokaryotic Argonaute (pAgo) genes were found mostly in archaeal MAGs at elevated temperatures up to 81.8°C. Overall defense gene repertoire declines downcore, as temperature increases and phylogenetic host range narrows, with phylogeny as the decisive control factor. We suggest that these defense systems, together with DNA repair mechanisms, protein maintenance activities, and RNA modification pathways, constitute a survival toolkit for the hydrothermally influenced subsurface, where energy limitation and temperature extremes select for resilient microbial communities.
|
|
Scooped by
mhryu@live.com
July 12, 1:37 PM
|
RNA inverse folding, the design of RNA sequences that fold into specified target structures, is a central problem in RNA design, with applications in functional RNA engineering, synthetic biology, and nucleic-acid therapeutics. This task becomes especially challenging for pseudoknotted target structures because pseudoknots disrupt the nested structure assumed by standard thermodynamic folding models. Existing pseudoknot inverse-folding methods often rely on structure-predictor-based objectives. Direct optimization of the thermodynamic folding probability of a specified pseudoknotted target remains limited. This requires an evaluator that can assign target-specific folding probabilities within a pseudoknot-aware ensemble and can be used as an optimization signal. Results: We present PKProbDesign, a sampling-based inverse-folding framework that directly optimizes a thermodynamic folding-probability objective for pseudoknotted targets. For each target, candidate sequences are scored by combining the folding probability of a pseudoknot-free scaffold with the conditional folding probability of the remaining extension component. On 354 PseudoBase++ targets, PKProbDesign achieved the highest folding probability on 221 targets, compared with 117 for DesiRNA and 16 for MODENA. PKProbDesign demonstrates that pseudoknot inverse folding can be formulated around target folding probabilities rather than structure-prediction agreement alone. By combining scaffold decomposition with HFold/CParty-consistent conditional-ensemble evaluation, the method provides a practical probability-based framework for designing sequences for density-2 pseudoknotted targets. Availability: The source code of PKProbDesign is available at https://github.com/TakumiOtagaki/ PKProbDesign.
|
|
Scooped by
mhryu@live.com
July 12, 1:23 PM
|
CRISPR-based biosensing has rapidly emerged as a promising platform for environmental monitoring due to its high specificity, programmability, and compatibility with portable readouts. However, translation from biomedical diagnostics to environmental matrices remains challenging because of diverse sample types, complex inhibitors, and the breadth of biological and chemical targets. This Review provides a comprehensive analysis of CRISPR-based sensing technologies tailored for environmental contaminant detection, spanning both biological and chemical targets. We systematically evaluate published studies across target classes, Cas effectors, recognition mediators, sample matrices, pretreatment strategies, preamplification or signal-gain approaches, readout modalities, and reported performance metrics. To support practical implementation, we summarize a five-step experimental framework for environmental CRISPR sensing. We then propose a decision-guided design flowchart that links monitoring goals and matrix constraints to the selection of effectors, mediator-enabled transduction routes, pretreatment modules, amplification strategies, readouts, and validation controls. We further benchmark reported detection limits by normalizing units and comparing trends across preamplification-aided versus preamplification-free designs and by contextualizing performance against relevant regulatory or guideline thresholds when available. Across the literature, most studies rely on spiked-matrix validation, highlighting the need for broader nonspiked real environmental sample testing and more transparent reporting of sampling, pretreatment, and performance evaluation. Finally, we advocate standardized data reporting, including consistent units, workflow metadata, and matrix-matched validation, to enable cross-study comparison and accelerate the deployment of CRISPR-based sensors for real-world environmental monitoring.
|
|
|
Scooped by
mhryu@live.com
Today, 1:40 AM
|
The use of nitrogen (N) fertilizers to meet global food demands is expected to continue rising. However, up to 70% of N applied to agricultural soils is lost through microbially mediated processes such as nitrification. Inhibiting nitrification is thus a key strategy to reduce N losses and improve fertilizer N use efficiency. Various plant-derived compounds, termed biological nitrification inhibitors (BNIs), have been shown to reduce accumulation of nitrification products, intermediates, and byproducts (nitrite, nitrate, nitric and nitrous oxides). However, the mechanisms by which BNIs affect nitrifiers, along with their specificity and persistence in soil are not well understood. Here, we evaluated the effects of three BNIs: methyl 3-(4-hydroxyphenyl) acrylate (MHPA), 6-methoxy-2(3H)-benzoxazolone (MBOA), and limonene, on ammonia-oxidizing, total microbial, and fungal communities in two soils with contrasting pH. Their persistence in each soil was also evaluated. Although ammonia-oxidizing archaea initially dominated nitrifier communities in both soils, their bacterial counterparts significantly increased after mineral N addition but also were more sensitive to BNI application. Limonene and the synthetic inhibitor DMPP stimulated ammonium immobilization, as total soil mineral N was significantly reduced. Limonene and MHPA had the strongest off-target effects, increasing the relative abundance of hydrocarbon-degrading bacteria and potential fungal pathogens, respectively. In contrast, MBOA inhibited nitrification with minimal off-target effects. Among the tested BNIs, MBOA was also the most persistent in the high-pH, high-nitrification-rate soil. Our results show that MBOA is a promising biological inhibitor and highlight the importance of understanding BNIs' ecological effects to develop targeted and sustainable N management strategies.
|
|
Scooped by
mhryu@live.com
Today, 1:04 AM
|
Novel genes arise through multiple mechanisms, including gene duplication, gene fusion, and horizontal gene transfer (HGT). While HGT has increasingly been documented in animals, the posttransfer evolutionary fate of horizontally acquired genes is less well understood. We hypothesized that fusion with endogenous sequences in animal genomes might generate what we call “HGT-chimeras”: genes with regions of nonmetazoan and metazoan descent in the same open reading frame. To test this hypothesis, we developed a molecular phylogenetics pipeline that enables the identification of HGT-chimeras. We applied our pipeline to 319 high-quality annotated arthropod genomes and uncovered a high-confidence set of 274 HGT-chimeras corresponding to 104 independent origination events across diverse arthropods. HGT-chimeras contain intervals acquired from across the tree of life, and many likely originated via a gene duplication-based mechanism. To assess whether HGT-chimeras might be functionally important, we performed RT-PCR and Sanger sequencing of tissues from 20 arthropod species predicted to harbor HGT-chimeras in their genome. We found evidence for the expression of contiguous chimeric messenger RNA transcripts (mRNAs) for 36 of 41 tested HGT-chimeras across 18 of 20 different tested species. We also found evidence that HGT-chimeras evolve under purifying selection and have acquired potentially functional domain architectures, consistent with the hypothesis that these genes are in active use and may participate in diverse biological processes. These results illuminate an underappreciated combinatorial mechanism underlying the origin of novel genes across the largest animal phylum, and suggest that interdomain sequence fusion can play important roles in animal biology and evolution.
|
|
Scooped by
mhryu@live.com
Today, 12:27 AM
|
We test the idea that innate agricultural soil microbiomes can be engineered to help support crops under climate change conditions. Salinization is one of the key drivers of agricultural soil degradation globally, and rising sea levels combined with decreasing rainfall are exacerbating this threat to food production. Changes in soil microbial community structures measured from DNA and functions and fitness measured from RNA and labeled amino acid uptakes support the hypothesis that the deliberate use of part-saline irrigation adapts soil microbiomes to increased salinities. While saline irrigation suppressed crop establishment in some cases, this microbiome response combined with inferences of increased microbial nutrient cycling and energy management correlates with final crop yield data and supports the hypothesis that engineered soil communities have helped protect yields under increased salinity conditions. This work provides evidence of the efficacy of a novel pragmatic, cost-effective innate soil microbiome engineering intervention for optimizing and securing food systems for future climate change conditions.
|
|
Scooped by
mhryu@live.com
Today, 12:03 AM
|
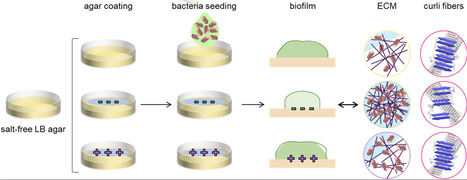
Biofilm extracellular matrix (ECM) varies with environmental conditions and substrate properties. Understanding the surface-biofilm relationship helps to perfect antibacterial strategies and to design new engineered living materials (ELMs). In this work, we studied how cationic and anionic polyelectrolyte coatings affect macroscopic features of E coli curli-producing biofilms, as well as the properties of their curli amyloid fibers. Cationic coatings limited biofilm spreading, increased their surface density and water absorption, which correlated with a higher yield of curli amyloid fibers with looser structure. In contrast, anionic surfaces allowed for standard biofilm spreading, with a lower fiber yield but a more compact and chemically stable fiber structure. Higher biofilm rigidity and adhesion were measured on both types of charged surfaces. Thus, we propose that the differences in biofilm macroscopic properties result from a trade-off between curli quantity and quality in the ECM, namely fiber density and molecular packing, as well as their interaction with water. Our findings provide insights on how the biophysical properties of the ECM can be controlled by tuning the substrate physico-chemical characteristics with charged coatings. This work opens up new avenues for developing antimicrobial strategies, as well as tailoring the properties of amyloid-based ELMs.
|
|
Scooped by
mhryu@live.com
July 13, 11:04 PM
|
Synthetic biology has recently proven to be a valuable tool for enhancing agriculture even in the face of environmental and biological stresses. Understanding the challenges that militate synthetic biology will assist in ensuring safe, stable, and scalable crop production. Thus, we examined the challenges limiting synthetic biology applications in engineering plant–microbe partnerships and highlighted future research directions. Ecological, biological, technical, and regulatory barriers to synthetic biology-driven plant–microbe engineering are the challenges examined in this review. Profound insight into these challenges will lead to a shift toward systems-level approaches that integrate multiomics analyses, predictive modeling, and framework-responsive genetic designs. To completely translate synthetic biology from the laboratory to the field, improved delivery methods, monitoring strategies, and harmonized regulatory frameworks should be encouraged. In addition, the development of robust and controllable microbial chassis should be emphasized. syncom
|
|
Scooped by
mhryu@live.com
July 13, 10:56 PM
|
Engineered bacterial routes for oxidation of non-native alcohols face three challenges: Nicotinamide-dependent enzymes are coupled to cellular redox metabolism, nicotinamide-independent aryl-alcohol oxidases (AAOs) usually express poorly in bacteria, and aldehyde products are rapidly modified by host enzymes. Here, we address these limitations by engineering aldehyde-retaining E. coli for discovery and application of soluble bacterial AAOs. Screening 51 candidates revealed a high-expression sequence cluster containing enzymes that are active on diverse aromatic and furan-based alcohols. Pairing the top-performing AAO with designer pathways in aldehyde-retaining cells enabled modular C-N and C-C bond forming cascades starting from supplied alcohols. By making both the oxidase and its product compatible with the host, this work advances air-driven oxidation of diverse alcohols as a programmable entry point to aldehyde-derived chemistry in engineered bacteria.
|
|
Scooped by
mhryu@live.com
July 13, 10:33 PM
|
Protein function prediction traditionally relies on structured gene ontology (GO) labels or multi-label classifiers. However, these labels or classifiers cannot flexibly describe molecular function, biological process, cellular component, and free-text functional narratives in a single output. In comparison, generation-based approaches offer an intuitive paradigm for flexible free-text protein annotation, with large language models (LLMs) as a representative method for protein-text modeling. Recent efforts on utilizing LLMs for protein semantic understanding and annotation generation have adopted sequence-only encoding or sequence-text contrastive alignment paradigms, yet without explicit consideration of three-dimensional structural information. To address these limitations in current protein function prediction methods, we present ProtBLIP2-SST, a two-stage framework built on the BLIP2 model architecture that bridges protein sequence, structure, and text for open-ended protein functional caption generation. Specifically, we first integrate sequence and structure information through SaProt, a protein language model (PLM) with a structure-aware vocabulary that fuses residue tokens with Foldseek-derived 3Di structural tokens. To empower the LLM to understand protein semantics, we employ a Q-Former (a querying transformer in BLIP2) with learnable query tokens as the cross-modal projector to align protein features from the frozen SaProt encoder and text features from a frozen BiomedBERT via protein--text contrasting, protein--text matching, and protein captioning objectives. After alignment, the protein features are linearly projected and prepended to the prompt embeddings of the LLM for protein captioning fine-tuning with LoRA. Trained on 441k protein--text pairs from Swiss-Prot with corresponding structures from the AlphaFold Database, our ProtBLIP2-SST outperforms sequence-only and sequence-text alignment baselines on protein captioning metrics, with ablation studies demonstrating the effectiveness of integrating structure with sequence information for improved protein understanding. Through a unified two-stage alignment-and-generation pipeline, ProtBLIP2-SST integrates protein sequence and structural information, overcomes the rigidity of traditional GO-centric classification, generating open-ended captions that jointly describe molecular function, subcellular location, and homology context in one single output.
|
|
Scooped by
mhryu@live.com
July 13, 10:12 PM
|
Predicting the location of metal-binding sites in proteins is crucial for fundamental biological questions and biotechnological applications. Over the past decade, the rise in metal-bound protein structures in the Protein Data Bank, combined with advanced statistical models such as deep learning, has accelerated the development of metal-binding site prediction tools. Several approaches are now available, offering high-quality benchmarks and predictive performance. Our initial development in this area is BioMetAll, whose first version was based on backbone pre-organization. Here, we introduce its second version, featuring two major updates: 1) metal-specific scoring functions and 2) prediction using backbone geometry alone or in combination with first coordination sphere descriptors. Apart from demonstrating metal sensitivity and yielding better benchmarking results, this new version allows the assessment of the influence of considering the metal first coordination sphere versus backbone pre-organization on how metallic species bind to proteins.
|
|
Scooped by
mhryu@live.com
July 13, 5:24 PM
|
Targeted protein degradation (TPD) is an emerging therapeutic modality, but how multiple factors jointly determine degradation efficacy remains poorly understood. Here we present a quantitative framework for modeling the cellular mechanism of action of TPD, allowing for prediction of cellular efficacy with practically obtainable parameters. Applying this model to published data on 41 targets reveals a common range of degradation rate between 0.1 and 10 min−1, reflecting a characteristic efficiency of the TPD process. We define the degradability landscape, which provides a holistic characterization of the degradation propensity of a target of interest and serves as a roadmap for degrader optimization. We show how degradability is influenced by key factors such as target half-life and E3 level. We further quantify functional inhibition for kinase targets, enabling direct comparison between degrader and small molecule inhibitors. Finally, we uncover degrader discovery opportunities by systematically identifying targets with the potential to achieve superior degradation to facilitate TPD translational development. Targeted protein degradation is an emerging therapeutic modality, but degradation efficacy remains unpredictable. Here, the authors present a quantitative framework that predicts cellular degradation efficacy, and shows how it can guide target selection and degrader optimization.
|
|
Scooped by
mhryu@live.com
July 13, 2:41 PM
|
Although probiotic-based bionic strategies show therapeutic promise for inflammatory bowel disease, their clinical translation is limited by poor gastric acid survival, inefficient intestinal colonization and inadequate targeting. Inspired by the multi-level cooperative mechanism of defense protection–danger sensing–tissue repair observed in coral communities, we developed a core–shell bionic microcapsule reactor (MY-E@SS). Here we show that the multifunctional bionic shell enables safe delivery of engineered bacteria through the gastrointestinal tract. Upon reaching inflamed intestinal sites, these bacteria sense the pathological microenvironment and responsively release an anti-inflammatory peptide. In a male murine model of inflammatory bowel disease, this system exhibited excellent biocompatibility and pronounced therapeutic efficacy, restoring intestinal barrier integrity, attenuating systemic inflammation and oxidative stress, modulating respiratory metabolism, and reestablishing microbial homeostasis. Mechanistically, therapeutic effects were attributed to inhibition of TNF-α/NF-κB signaling pathway. This work provides an intelligent platform to modulate inflammatory microenvironments and advance therapies for complex diseases. The authors developed a bionic microcapsule containing engineered bacteria that sense gut inflammation and release targeted therapy. Microcapsule treatment in mouse model of inflammatory bowel disease restored intestinal barrier function.
|
|
Scooped by
mhryu@live.com
July 12, 1:39 PM
|
Proteins of unknown function represent a significant gap in our understanding of biological processes, encompassing large portions of the proteomes of many organisms, especially prokaryotes. Addressing this gap is critical to understanding the biology and pathogenicity of such organisms. We introduce ProtPen, an open-source pipeline that facilitates protein function prediction by combining eggNOG-mapper for sequence-based annotation with Foldseek for rapid structural similarity searches using AlphaFold-predicted protein structures. Annotation results from both tools are merged and enriched with UniProt metadata to produce a comprehensive output suitable for downstream analysis. The pipeline requires only a FASTA input file with UniProt identifiers, and is designed to analyze datasets on the scale of whole proteomes. Benchmarking on a curated dataset of well-characterized Pseudomonas aeruginosa proteins demonstrated an annotation accuracy of >90%, and highlighted the complementarity of sequence- and structure-based methods. Further evaluation of ProtPen included its application to biologically relevant datasets, comprising proteins of unknown function that exhibited significant differential abundances in a proteomics dataset of P. aeruginosa, and uncharacterized glycoproteins from Haloferax volcanii. ProtPen is readily extensible to incorporate additional protein function prediction tools. In summary, this pipeline facilitates the systemwide annotation of proteins of unknown function from proteomic datasets and whole proteomes.
|
|
Scooped by
mhryu@live.com
July 12, 1:27 PM
|
Large language models (LLMs) are deep learning-based artificial intelligence models that have achieved remarkable success in natural language processing. Typically composed of neural networks with billions of parameters, they are trained on massive unlabeled datasets using self-supervised or semi-supervised learning. Beyond language, LLMs hold immense potential for addressing complex bioinformatics challenges. This review provides a comprehensive overview of transformer-based model applications in genomics, transcriptomics, proteomics, drug discovery, and single-cell analysis. We discuss critical components, including tokenization strategies for diverse biological data, transformer architectures, attention mechanisms, and pretraining approaches. We also survey currently available foundation models and their downstream applications across bioinformatics domains. Finally, we highlight major challenges that remain insufficiently addressed in prior reviews and outline future perspectives and design principles for next-generation biological language models, offering practical guidance for both users and developers.
|