Your new post is loading...

|
Scooped by
mhryu@live.com
Today, 3:31 PM
|
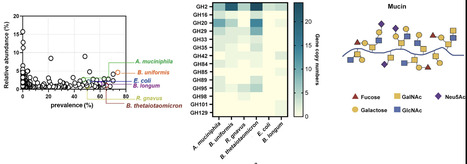
The gut microbiota is a complex microbial community that plays a crucial role in host health. Environmental and biological factors within the ecosystem influence the dynamic interactions among its members. Although dietary and host-derived nutrient availability play a key role in shaping microbial ecology and interaction patterns, the dynamics of these interactions within the mucus layer remain poorly understood. In this study, we analyzed a synthetic community comprised of six species with variable abilities to utilize mucin. We performed in vitro growth analyses and monitored the interactions among community members in monoculture, co-culture, and community batch culture under different nutrient conditions. Our results showed that positive interactions were prevalent among bacteria when mucin served as the sole carbon source. In contrast, the addition of glucose or high nutrient availability significantly increased inter-bacterial competition. These findings suggest that mucin mitigates competitive antagonism and potentially promotes community diversity. Further in vivo studies supported the role of mucin in increasing community diversity and modulating bacterial metabolic patterns. Deciphering these intricate relationships is essential for understanding how gut microbiota stability is maintained, and what factors might disrupt this delicate balance.
|
|
Scooped by
mhryu@live.com
Today, 3:17 PM
|
The Modular Cloning (MoClo) and PhytoBrick standards have revolutionized plant synthetic biology by establishing a standardized, hierarchical assembly grammar. However, as the engineering of complex metabolic pathways, multi-trait stacks, and synthetic gene circuits expands, existing toolkits hit practical boundaries in assembly capacity and fixed grammars. To overcome these bottlenecks, we present MozClo, an expansion of the MoClo/PhytoBrick architecture. MozClo expands the standard Level 1 assembly framework to 10 positions using new L1 acceptors, end-linkers and dummy parts. We also identify and resolve a critical, sticky-end collision at L1 position 7 that has caused assembly failures during L2 cloning of large plasmids. To address commercial DNA synthesis length constraints and to lower cloning costs, we designed a universal 5-in-1 gene fragment multiplexing system. This architecture embeds up to five distinct parts flanked by orthogonal pairs of BpiI restriction sites into a single synthesized fragment, allowing them to sort independently into their respective L0 acceptor plasmids while maintaining complete modular flexibility of part types. Finally, we provide Level 2 cloning backbones with built in selection genes for common soybean transformation methods to facilitate downstream plant selection. Together, these advancements reduce DNA synthesis overhead and accelerate the construction of complex multigene payloads for plant biotechnology.
|
|
Scooped by
mhryu@live.com
Today, 3:10 PM
|
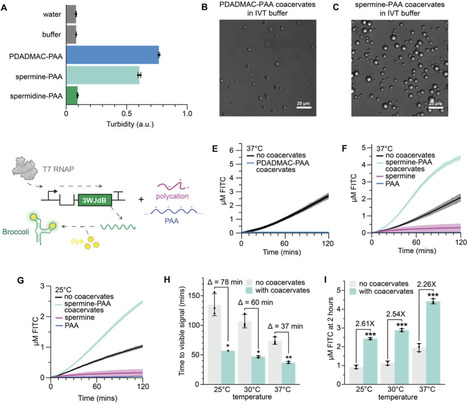
Cell-free biosensors leverage in vitro gene expression reactions to detect chemicals. While inexpensive, modular, and distributable, these platforms are constrained by slow readouts at ambient temperatures, precluding practical field operation. In cells, phase separation accelerates biochemical reactions; however, recapitulating these gains in vitro has remained challenging for complex biochemistries. Here, we report the first self-assembling coacervate system that accelerates in vitro transcription. Prepared by simple mixing, coacervation with spermine and polyacrylic acid occurs dynamically in response to NTP consumption and co-localizes DNA templates and RNA polymerase to accelerate transcription, mimicking intracellular phenomena. We exploit this discovery to accelerate the cell-free biosensing of six ligands, demonstrating that coacervation can preserve platform modularity, improve sensitivity, retain lyophilization compatibility, function in field matrices, and reduce ambient-temperature time-to-signal by hours. This work contributes to a growing understanding of phase separation in biology and advances the use of membrane-less organization for real-world applications.
|
|
Scooped by
mhryu@live.com
Today, 2:51 PM
|
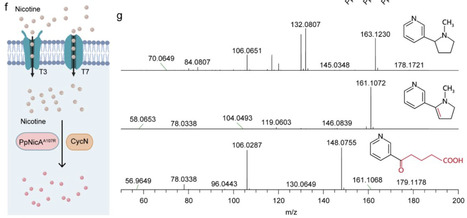
Nicotine accumulates in the gut and drives non-alcoholic steatohepatitis (NASH) via the gut-liver axis, yet no effective clinical intervention is currently available. To address this challenge, the probiotic E. coli Nissle 1917 (EcN) was engineered for in situ nicotine clearance in the gut. Mutational screening of nicotine oxidoreductase 2 (PpNicA2) identified a highly active variant, PpNicA2A107R. Its incorporation into EcN together with an electron transfer protein (CycN) and a newly identified transporter (T3/T7) yielded 80% nicotine-degrading activity. Chromosomal integration of this module generated a stable strain, EcN-N12, which in NASH mouse models depleted intestinal nicotine, rescued hepatic lipid metabolism, alleviated tissue damage, and intercepted the nicotine-mediated gut-liver axis pathological progression. This work thus offers an effective and clinically translatable approach for nicotine-associated diseases.
|
|
Scooped by
mhryu@live.com
Today, 2:31 PM
|
Small RNAs (sRNAs) mediate gene silencing through RNA interference (RNAi). These sRNAs include small interfering RNAs (siRNAs) and microRNAs (miRNAs) that regulate gene expression and immunity in plant–microbe interactions. Cross-kingdom RNA interference (ckRNAi) refers to RNAi between organisms belonging to different kingdoms of life. A well-studied example of ckRNAi is the bidirectional exchange of sRNAs between plants and their pathogens where sRNAs silence host defence genes or pathogen virulence factors. Thus, ckRNAi is part of the molecular dialogue between plants and associated microbes. Pioneering studies indicated that fungal pathogens such as Botrytis cinerea deliver sRNAs to hijack the host silencing machinery and to suppress immunity, while plants, in turn, export sRNAs to silence the virulence genes of pathogens. ckRNAi has lately been found in symbiotic interactions, that is in arbuscular mycorrhizas and ectomycorrhizas. In arbuscular mycorrhizas, fungal sRNAs from Rhizophagus irregularis target host transcription factors that restrict symbiosis, thereby facilitating fungal accommodation. Studies have also demonstrated that bidirectional RNA exchange enhances symbiotic efficiency by modulating gene expression in both partners. The ectomycorrhizal fungus Pisolithus microcarpus deploys a symbiosis-induced miRNA in colonisation of Eucalyptus grandis. Therefore, sRNAs regulate host transcriptional networks in both mutualistic and pathogenic plant–fungal relations. The study (2026; doi: 10.1111/nph.71187), represents a further expansion of this paradigm. An essential finding of this work is that the characterisation of sRNA-based communication within the tripartite interaction between Arabidopsis thaliana, the fungus Trichoderma atroviride, and the fungal pathogen B. cinerea. Trichoderma atroviride has plant-beneficial effects by protecting plants from pathogens. The molecular mechanisms underlying this protective effect have, however, remained largely elusive.
|
|
Scooped by
mhryu@live.com
Today, 1:20 AM
|
Profiling microbiomes is an important way to understand the function and composition of communities in the wild, but natural microbiomes are often highly complex and often unamendable to experimentation to reveal cause and effect relationships. By using a small group of cultivable strains to represent those found in the wild, synthetic communities are one solution to this problem. Here we describe the MAize Rhizosphere Synthetic Community (MARSc), a genome-enabled 31-member bacterial community representative of the diversity found on the roots of maize grown in Iowa soils. This community is built around Pseudomonas putida KT2440, a model maize rhizosphere colonist and synthetic biology chassis. We characterized microbe-microbe interactions and biofilm formation of MARSc members in a variety of environmental contexts, finding that both behaviors are broadly controlled by nutrient levels. Genomic analysis and microbiome profiling of these organisms revealed that annotated biofilm genes (such as surface attachment and exopolysaccharide production) correlated to rhizosphere colonization, but neither trait correlated to in vitro biofilm formation. In vitro interactions assay findings were surprisingly consistent with co-correlations of rhizosphere abundance amongst MARSc members on roots. Finally, we found that when applied to the roots, MARSc can increase maize growth under nitrogen-limiting conditions. Altogether, MARSc is a useful tool for identifying some of the factors influencing rhizosphere microbiome assembly and will be a strong foundation for further work in this area.
|
|
Scooped by
mhryu@live.com
Today, 1:11 AM
|
Mechanistic ordinary differential equation models are widely used in systems biology to represent biochemical networks, population dynamics, cell-state transitions, and other biological processes; however, their predictive value depends critically on accurate parameter estimation from noisy and often sparse experimental data. In this tutorial, we present the Weak-form Estimation of Nonlinear Dynamics (WENDy) method as a forward-solver-free approach that reformulates parameter estimation as a covariance-corrected weak-form regression problem by integrating the model equations against compactly supported test functions. We present the background on the methodology through the lens of the familiar logistic equation, and we demonstrate applications of the method on real experimental data through two systems biology examples: a glycolytic oscillator with relatively dense time-course data and a sparse epithelial-mesenchymal cellstate transition model with multiple experimental replicates. Ultimately, using WENDy, we estimate interpretable biological parameters with uncertainty for systems with noisy and sometimes sparse available experimental data.
|
|
Scooped by
mhryu@live.com
Today, 1:06 AM
|
Bacterial bioremediation involves bacterial strains and communities and their complex interactions with the environment, aiming to restore ecological balance by degrading contaminants in natural systems (soil, water, air). These processes rely on coordinated gene clusters encoding catabolic pathways. Many xenobiotic-catabolic gene clusters (XGCs) reside on mobile genetic elements (MGE), enabling horizontal gene transfer (HGT) and genome rearrangements that drive rapid microbial adaptation to anthropogenic contaminants. Here we review the evolutionary and ecological roles of HGT and genome restructuring in assembling and optimizing biodegradative functions. We introduce the concept of metabolic HGT hubs—microbial taxa, mobile elements, and ecological features that serve as central nodes for gene exchange—facilitating metabolic innovation and cooperation within microbial consortia. These processes enhance ecosystem resilience and pollutant degradation efficiency by promoting functional redundancy and metabolic division of labor. Understanding these dynamics informs strategies for engineering microbial communities and genetic bioaugmentation to improve bioremediation outcomes. Our perspective highlights bioremediation as an extension of metabolic network evolution under anthropogenic selection, emphasising both its potential and the need to consider ecological and biosafety implications.
|
|
Scooped by
mhryu@live.com
Today, 12:41 AM
|
Programmable molecular biology increasingly requires strategies for converting engineered recognition or proximity modules into measurable outputs, particularly within transcriptional regulation, RNA imaging, and CRISPR-associated systems. Synthetic chemically induced dimerization (CID) systems provide a class of programmable recognition modules for such applications, yet generalized strategies for coupling structurally diverse CIDs to functional readouts remain limited. Here, we introduce a CID-to-output conversion strategy based on engineering of the linker-mediated coupling interface. Using single-fluorescent-protein sensors as an experimentally tractable optical model readout, we systematically varied paired N- and C-terminal linkers flanking circularly permuted green fluorescent protein (cpGFP) to map coupling landscapes across synthetic CID systems derived from combinatorial selection and computational protein design. The results revealed strong non-additive interactions across paired linkers and suggest that linker length is a first-order determinant of CID-to-output coupling. Across nanobody-, monobody-, and de novo-designed CID architectures, this framework yielded functional sensors with dynamic ranges up to 1270% and robust responses in mammalian cells. Together, this work demonstrates that effective CID-to-output conversion can be achieved by empirically mapping the linker-mediated coupling interface, providing a practical route for adapting synthetic CID to diverse programmable molecular readouts and nucleic-acid-associated synthetic biology systems
|
|
Scooped by
mhryu@live.com
Today, 12:35 AM
|
Protein-quality-control systems are essential for cells to maintain protein homeostasis during both steady-state growth and acute stress. Yet, how individual chaperone molecules dynamically reorganize in living cells as protein unfolding and aggregation rates change, remains poorly understood. Here, we use super-resolution imaging and single-molecule tracking to define the in vivo dynamics of the bacterial heat shock protein 70 (Hsp70) DnaK in live E. coli. We found that majority of DnaK molecules actively engage with the proteome already at the optimal growth temperature, while heat stress induces the accumulation of a distinct slow-moving DnaK population with increased dwell times. These interactions occupy different intracellular regions, suggesting spatially separated chaperone activities. Finally, DnaK folding activity becomes burdened in cells lacking co-chaperones, such as IbpAB, HtpG or DnaJ. Overall, our findings reveal transient proteome interactions as a main chaperone mode of action, enabling DnaK to sense and mitigate proteotoxic stress in living cells.
|
|
Scooped by
mhryu@live.com
July 9, 11:41 PM
|
Currently, fossil fuels are the main and dominant sources for producing fuels and commodity chemicals. The rising demand for fossil fuels continues to entrench global dependency on non-renewable sources creating the climate risk, the economic instability and sustainability challenges. Hence, there is a need to shift towards globally available, sustainable and renewable resources such as lignocellulosic biomass (LCB) that offers a sustainable pathway for producing biofuels and chemicals. Though various technologies are available for LCB conversion to biofuels and chemicals, scaling up of biorefineries remains stifled by its recalcitrance nature and volatile supply chain economics. LCB processing often requires pretreatment to disrupt the rigid lignin–hemicellulose barrier and decrystallize cellulose. This structural opening is essential to maximize the enzymatic hydrolysis and sugar yields for biofuel production. The pretreatment process is energy-intensive and expensive accounting for 40% of the overall biofuel production cost followed by hydrolysis using expensive enzyme cocktails. These economic barriers currently limit the adoption of LCB as a cost-competitive fuel resource. The promising strategy for low cost LCB derived ethanol production is to adopt integrated biorefinery approach utilizing physical, chemical and biological processes. The integrated biorefinery approach tackles the high costs of second generation ethanol production by mimicking traditional petroleum refineries. It valorizes all three LCB components to marketable fuels and high-value chemicals maximizing the overall process profitability. This review discusses on the latest developments in biofuels production processes especially in relation to ethanol and butanol production.
|
|
Scooped by
mhryu@live.com
July 9, 11:10 PM
|
"Aerobic glycolysis” is a widely used term whose current meaning has drifted from its original usage in a way that has created confusion and inaccuracy. This drift has weakened “aerobic glycolysis” as a hypothesis-testing framework, despite the critical importance of glycolysis in understanding cellular bioenergetic behavior. Here, we examine the historical and contemporary uses of “aerobic glycolysis” and the related “Warburg effect”. We argue that “aerobic glycolysis” as originally investigated was essentially a bioenergetic phenomenon. We review the bioenergetic model of glycolysis and mitochondrial respiration as ATP supply pathways operating together to meet cellular ATP demand. A bioenergetic view of aerobic glycolysis clarifies that it is not a less desirable contingency or indicator of pathology, but rather a part of a kinetically regulated system of cellular energy supply. On this basis, the operation of glycolysis under many different physiological and pathological conditions can be better interrogated and understood. This Perspective examines how “aerobic glycolysis” originally described a bioenergetic system behavior. Reintroducing a bioenergetic conceptual framework to this term could strengthen its meaning and resolve confusion in the field.
|
|
Scooped by
mhryu@live.com
July 9, 10:57 PM
|
Millions of phage genomes have been mined from metagenomic data recently but the genome completeness remains poor because of the limitations of existing phage detection methods, which rely on metagenomic contigs that fragment phage genomes. Here, we present PALACE, a conjugate-graph-based framework for assembling high-quality phage genomes from metagenomes. PALACE incorporates homology-based and deep-learning-based methods to detect phage signals and constructs a conjugate graph from the metagenomic sample. On simulated data, PALACE generates accurate and complete phage genomes, achieving an F1 score of 0.92–1.00 across simulation settings, outperforming the second-best method by 0.21–0.48. Applying PALACE to 914 gut metagenomic samples from healthy controls and participants with colorectal cancer (CRC) yielded 5,306 high-quality phage genomes, outperforming the second-best benchmark method by 55.98% in median genome completeness. We observed a high degree of functional organization for genes within phage genomes. Phages from participants with CRC exhibited a notable enrichment of metabolic factors, suggesting their adaptation to nutrient availability in the CRC gut environment. PALACE combines homology features and deep learning to improve phage assembly from metagenomics.
|
|
|
Scooped by
mhryu@live.com
Today, 3:24 PM
|
The human microbiome is inherently structured by phylogeny, yet most predictive models treat microbial taxa as independent features, thereby underusing evolutionary information that may improve disease classification. While recent deep learning approaches have attempted to incorporate phylogeny, they generally rely on projecting phylogenetic trees into Euclidean spaces, which can distort the intrinsic topology of evolutionary relationships. To address this limitation, we propose PhyloGCNE, a framework that models microbiome samples directly as graphs and employs edge-aware graph convolution to integrate phylogeny. Unlike previous methods that rely on fixed, distance-based aggregation, PhyloGCNE learns how phylogeny-informed edge attributes should influence signal propagation across evolutionary hierarchies. We further introduce a Phylogenetic Saliency Propagation (PSP) framework for model interpretation, which attributes importance scores to microbial taxa by integrating gradient sensitivity with evolutionary context. Benchmarked against one synthetic and eight real-world data sets spanning inflammatory bowel disease, colorectal cancer, type 2 diabetes, oral squamous cell carcinoma, gastric cancer, and dietary fiber intervention, PhyloGCNE consistently outperforms existing state-of-the-art approaches. Together, these results establish PhyloGCNE as an accurate and interpretable phylogeny-aware framework for microbiome-based host phenotype prediction.
|
|
Scooped by
mhryu@live.com
Today, 3:15 PM
|
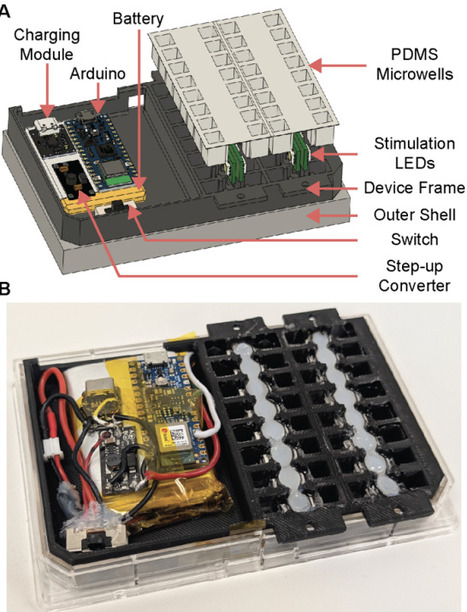
Optogenetic control enables light-actuated regulation of gene expression and provides a programmable interface between living cells and electronic systems. However, routine prototyping of optogenetic constructs remains limited by infrastructure. Existing closed-loop platforms often require chemostats, microfluidics, robotic handling, or custom optical sensors, which can increase cost, reduce accessibility, or constrain measurement performance. Here, we present LEMOS 2.0, an updated LED-Embedded Microplate for Optogenetic Studies, a low-cost device for optogenetic stimulation and gene-circuit characterization inside standard off-the-shelf microplate readers. LEMOS 2.0 builds on the original LEMOS platform by increasing throughput from 16 to 32 microwells and reducing light leakage between adjacent microwells, allowing dark conditions to be used as an additional illumination state. The device consists of a 3D-printed frame, individually addressable LEDs positioned next to each microwell, a rechargeable battery, and an onboard microcontroller for Bluetooth-based wireless communication. Biocompatible polydimethylsiloxane microwells are cast directly into the device by replica molding, allowing bacterial cultures to be stimulated while optical density and fluorescence are measured by the microplate reader. This protocol describes the full LEMOS 2.0 workflow, including device fabrication, circuit assembly, Arduino programming, PDMS microwell casting, plate-reader setup, strain and culture preparation, automated experiment execution, device cleanup, and fluorescence/OD600 data analysis. As a demonstration, the protocol uses the CcaSR optogenetic system, in which sfGFP expression is activated by green light and repressed by red light. LEMOS 2.0 is intended to make optogenetic perturbation and gene-expression characterization more accessible to wet-lab users, enabling faster design-build-test-learn cycles without requiring specialized bioreactor or microfluidic infrastructure.
|
|
Scooped by
mhryu@live.com
Today, 3:04 PM
|
Adenine base editors (ABEs) enable efficient A•T to G•C conversion, but their broad activity windows frequently cause unintended bystander edits. We hypothesized that insertion of a bulky, inert protein domain into the base editor would limit the effective reach of the deaminase, thereby preferentially directing editing to the intended target adenine. Here, we systematically map domain insertion sites within the high-activity TadA8e adenine base editor using structure-guided design and computational inference. We find that TadA8e accepts domain insertions at multiple surface sites, with overall activity and editing window width dependent on insert position rather than domain identity. Excitingly, inserting domains at residue L68 preserved robust editing across multiple genomic loci while tightly focusing the editing window around position 5. Insertion of superfolder GFP at this site produced a base editor variant with a narrower editing window, the ability to track edited cells by fluorescence, and markedly reduced Cas-independent off-target editing. Our work highlights domain insertion engineering as a powerful strategy to create more focused and precise CRISPR base editors.
|
|
Scooped by
mhryu@live.com
Today, 2:34 PM
|
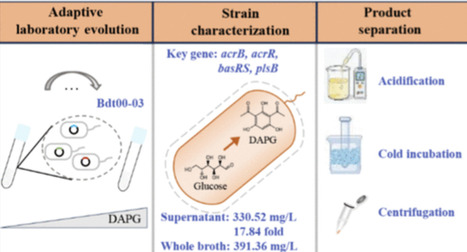
2,4-Diacetylphloroglucinol (DAPG) is a valuable antimicrobial compound with significant agricultural potential, suffers bioproduction limitations from host toxicity and inefficient downstream processing. This study engineered DAPG-hyper-tolerant E. coli via adaptive laboratory evolution (ALE) starting from a phloroglucinol-tolerant strain. Optimized shake-flask fermentation of evolved Bdt03 yielded 330.52 mg/L DAPG (17.84-fold of the wild type), and the yield from the whole fermentation broth could be further increased to 391.36 mg/L. A novel organic solvent-free extraction method recovered DAPG from fermentation broth with over 98% yield via acidification, cold incubation and centrifugation, simplifying downstream processing. Genomic resequencing identified several key mutations underlying DAPG tolerance, which were validated and stacked to precisely construct strain Bb03 with enhanced production and tolerance. This work addresses the critical bottlenecks in DAPG biosynthesis by enhancing host tolerance and developing a sustainable downstream processing strategy, and also offers valuable genetic insights for constructing high-yield DAPG-producing strains and advancing the application of DAPG-responsive genetic circuits in synthetic biology.
|
|
Scooped by
mhryu@live.com
Today, 2:05 PM
|
Hirschsprung disease (HSCR) is a complex developmental disorder of the enteric nervous system, primarily driven by regulatory variants within enhancer elements of the RET gene. To investigate how these variants lead to aganglionosis, we developed a humanized mouse model by inserting an intact 77kb human RET genomic locus into the Rosa26 safe-harbor locus. Utilizing “big DNA” synthetic biology and Bxb1-mediated recombination, we integrated the complete human locus including all exons, introns, and upstream regulatory elements which we validated via nanopore and short-read sequencing. Functional analysis confirmed in vivo human RET expression; however, our initial HSCR-associated “sensitive” haplotype expressed at only 21% of wild-type levels. This significant reduction proved insufficient to rescue the viability when endogenous mouse Ret was deleted. We identified that this deficiency is partially driven by five risk SNPs within established enhancers. Specifically, using CRISPR/Cas9 to restore a conserved So×10 binding site (converting a sensitive SNP to a protective one) increased RET expression by 1.9-fold and restored transcription factor binding. This study provides a robust framework for modeling human-specific regulatory disorders and demonstrates the critical impact of non-coding variation on disease pathogenesis.
|
|
Scooped by
mhryu@live.com
Today, 1:17 AM
|
CRISPR systems have revolutionized microbial genome editing. However, their reliance on DNA double-strand breaks (DSBs) and homologous recombination limits applications such as large DNA integration and engineering nonmodel microorganisms. CRISPR-associated transposase (CAST) systems provide a promising alternative, enabling DSB-free, recombination-independent, and programmable integration of large DNA fragments. This review summarizes the discovery and characterization of diverse CAST systems, the engineering strategies to enhance integration efficiency and specificity, and their applications in microbial engineering. Current limitations and future directions are also discussed. Overall, CAST systems hold great potential for advancing microbial genome editing, particularly in multi-copy DNA integration and genetic reprogramming of nonmodel microorganisms and complex microbial communities.
|
|
Scooped by
mhryu@live.com
Today, 1:09 AM
|
Recent advances in large-scale sequence mining have expanded our knowledge of RNA virus diversity. Most genome mining approaches for detecting RNA viruses rely on identifying the conserved RNA-dependent RNA polymerase (RdRp) by scanning sequencing datasets with specialized profile Hidden Markov Models (pHMMs). Recently, several new pHMM databases for RdRp detection have been released, each following distinct design principles. However, their relative performance remains unclear, and their accessibility to users without advanced computational expertise is limited. Here, we introduce the RdRp Collaborative Analysis Tool with Collections of pHMMs (RdRpCATCH: https://github.com/dimitris-karapliafis/RdRpCATCH), a platform that consolidates publicly available RdRp pHMM resources into a single, user-friendly framework. RdRpCATCH enables the scanning of (meta)transcriptomic assemblies to discover RNA viruses and provides subsequent taxonomic annotation of detected contigs. A comparative analysis of RdRp pHMM databases reveals that most are highly effective at detecting the known diversity of RNA viruses while minimizing false positives, supporting their joint use within RdRpCATCH. RdRpCATCH is distributed as both a conda package and a web server application (https://rdrpcatch.bioinformatics.nl), facilitating access for researchers with diverse levels of computational expertise. By integrating multiple pHMM resources, this unified framework addresses fragmentation in the field and reduces technical barriers, enabling comprehensive viral discovery.
|
|
Scooped by
mhryu@live.com
Today, 12:52 AM
|
Artificial intelligence is rapidly transforming everyday life and driving major advances across science. De novo enzyme design is an area of particular promise, with implications for medicine, biotechnology, and industry. Recent applications of AI-enhanced methodologies have yielded a range of enzymes with catalytic activities that were previously unattainable through conventional approaches. This perspective surveys emerging strategies and computational models that are redefining enzyme engineering and examines the opportunities and challenges on the path toward truly on-demand enzyme design.
|
|
Scooped by
mhryu@live.com
Today, 12:38 AM
|
The bacterial three-hybrid (B3H) assay is a powerful genetic tool for detecting interactions between RNA and RNA-binding proteins (RBPs) and assessing the consequences of RBP mutations. This transcription-based system connects the strength of an RNA–protein interaction to the expression of a lacZ reporter gene in E. coli cells. This in vivo approach allows researchers to dissect RNA-protein interactions within a cellular environment, bypassing the need for biochemical purification of RNAs or proteins. This chapter details a three-day protocol for generating quantitative B3H data. Since a significant challenge in B3H assays is RNA misfolding, we describe a recently optimized set of B3H constructs that mitigates this issue by isolating bait RNAs as discrete folding units.
|
|
Scooped by
mhryu@live.com
Today, 12:09 AM
|
Plant viruses, once viewed as harmful agricultural pathogens, are now powerful tools in biotechnology. Their nanoscale structure, self-assembly, and biocompatibility enable applications in agriculture, medicine, and environmental sustainability. They serve in gene delivery, genome editing, diagnostics, and nanomaterials for vaccines and drug delivery. Integration with AI, ML, and bioinformatics enhances virus discovery and prediction. Despite challenges, plant viruses are emerging as versatile, sustainable resources for global biotechnological innovations.
|
|
Scooped by
mhryu@live.com
July 9, 11:37 PM
|
Membrane-associated biomolecules, primarily proteins, are key enablers of communication, responsiveness, and complexity in natural living cells. Aiming to mimic these capabilities, there is growing interest in equipping bottom-up synthetic cells with membrane-associated biomolecular components. In this review, we focus on how proteins and nucleic acids have been associated with synthetic cell membranes, particularly lipid vesicles, to enable the transmission of signals across the membrane. We discuss strategies for anchoring these biomolecules into lipid bilayers and review how they can enable essential signalling mechanisms in synthetic cells, including cell tethering, the generation and fusion of vesicles, and signal transmission and transduction. We highlight how proteins offer native biological functionality, while nucleic acids may bring more modularity and control. Advancing this area will be essential for realising synthetic systems capable of studying natural communication mechanisms and unlocking applications in biosensing, therapeutics, and synthetic tissue engineering.
|
|
Scooped by
mhryu@live.com
July 9, 11:02 PM
|
Cells are the fundamental unit of life. Yet there is no natural cell for which all its life-essential functions are understood. Here we demonstrate a complete cell cycle for a synthetic cell undergoing selection, with genome replication, growth, resource acquisition via feeding, and genetically encoded division. The cell is encoded via a 90kb genome that includes functions needed for resource uptake, transcription, translation, growth, genome replication, and division. The resulting synthetic cell is sufficiently encouraging to support routinization of synthetic cell engineering workflows, and will ultimately underlie diverse applications across all of biotechnology.
|