 Your new post is loading...

|
Scooped by
mhryu@live.com
Today, 1:40 PM
|
Microbial defense systems are central to virus-host interactions and thus to microbial survival, but remain poorly studied in microbial communities of extreme environments. Here we examine the repertoire and distribution of microbial defense genes in hydrothermally influenced deep subsurface sediments and rocks of the Guaymas Basin (Gulf of California). Restriction–modification (RM) and abortive infection (Abi) systems were broadly distributed across the examined sediment depths, and CRISPR–Cas systems were primarily detected within temperate surficial sediments. Prokaryotic Argonaute (pAgo) genes were found mostly in archaeal MAGs at elevated temperatures up to 81.8°C. Overall defense gene repertoire declines downcore, as temperature increases and phylogenetic host range narrows, with phylogeny as the decisive control factor. We suggest that these defense systems, together with DNA repair mechanisms, protein maintenance activities, and RNA modification pathways, constitute a survival toolkit for the hydrothermally influenced subsurface, where energy limitation and temperature extremes select for resilient microbial communities.
|
|
Scooped by
mhryu@live.com
Today, 1:37 PM
|
RNA inverse folding, the design of RNA sequences that fold into specified target structures, is a central problem in RNA design, with applications in functional RNA engineering, synthetic biology, and nucleic-acid therapeutics. This task becomes especially challenging for pseudoknotted target structures because pseudoknots disrupt the nested structure assumed by standard thermodynamic folding models. Existing pseudoknot inverse-folding methods often rely on structure-predictor-based objectives. Direct optimization of the thermodynamic folding probability of a specified pseudoknotted target remains limited. This requires an evaluator that can assign target-specific folding probabilities within a pseudoknot-aware ensemble and can be used as an optimization signal. Results: We present PKProbDesign, a sampling-based inverse-folding framework that directly optimizes a thermodynamic folding-probability objective for pseudoknotted targets. For each target, candidate sequences are scored by combining the folding probability of a pseudoknot-free scaffold with the conditional folding probability of the remaining extension component. On 354 PseudoBase++ targets, PKProbDesign achieved the highest folding probability on 221 targets, compared with 117 for DesiRNA and 16 for MODENA. PKProbDesign demonstrates that pseudoknot inverse folding can be formulated around target folding probabilities rather than structure-prediction agreement alone. By combining scaffold decomposition with HFold/CParty-consistent conditional-ensemble evaluation, the method provides a practical probability-based framework for designing sequences for density-2 pseudoknotted targets. Availability: The source code of PKProbDesign is available at https://github.com/TakumiOtagaki/ PKProbDesign.
|
|
Scooped by
mhryu@live.com
Today, 1:23 PM
|
CRISPR-based biosensing has rapidly emerged as a promising platform for environmental monitoring due to its high specificity, programmability, and compatibility with portable readouts. However, translation from biomedical diagnostics to environmental matrices remains challenging because of diverse sample types, complex inhibitors, and the breadth of biological and chemical targets. This Review provides a comprehensive analysis of CRISPR-based sensing technologies tailored for environmental contaminant detection, spanning both biological and chemical targets. We systematically evaluate published studies across target classes, Cas effectors, recognition mediators, sample matrices, pretreatment strategies, preamplification or signal-gain approaches, readout modalities, and reported performance metrics. To support practical implementation, we summarize a five-step experimental framework for environmental CRISPR sensing. We then propose a decision-guided design flowchart that links monitoring goals and matrix constraints to the selection of effectors, mediator-enabled transduction routes, pretreatment modules, amplification strategies, readouts, and validation controls. We further benchmark reported detection limits by normalizing units and comparing trends across preamplification-aided versus preamplification-free designs and by contextualizing performance against relevant regulatory or guideline thresholds when available. Across the literature, most studies rely on spiked-matrix validation, highlighting the need for broader nonspiked real environmental sample testing and more transparent reporting of sampling, pretreatment, and performance evaluation. Finally, we advocate standardized data reporting, including consistent units, workflow metadata, and matrix-matched validation, to enable cross-study comparison and accelerate the deployment of CRISPR-based sensors for real-world environmental monitoring.
|
|
Scooped by
mhryu@live.com
Today, 1:13 PM
|
A plant-independent avenue for N2-fixation takes place in desert topsoils through a “C-for-N” mutualism between heterodiazotrophs and the cyanobacterium Microcoleus vaginatus. These partners come together within a diverse soil microbiome under conditions of N-limitation for the phototroph and C limitation for the heterotrophs. We hypothesized that extracellular chemical signaling might enable partner selection and collocation, though infomolecules shaping inter-microbial architecture were unknown. We show that the complex chemical composition of M. vaginatus’ exometabolome depends on its N-limitation status, thus potentially offering information to mutualists. In chemotactic assays, the exometabolome effectively repelled most native soil bacteria, particularly intensely when under N-limitation. Bacterial assemblages circumventing the repulsion were enriched in species that are rare in the soil microbiome, and that functionally resemble mutualistic cyanospheres (showing high N2-fixation potential, secretion of urea, and copiotrophy), setting the stage for a working symbiosis. Further, we could reproduce the enrichment of copiotrophs and nitrogen-fixers using mixtures of N-acetylglutamic acid, N-acetylmethionine, indole-3-acetic acid, and 5′-methylthioadenosine, all preferentially released by M. vaginatus under N-limitation. These signaling molecules did not result in an enrichment of urea producers, however. The results demonstrate that trans-species communication through specific infochemicals, together with already known quorum-sensing-like intraspecific communication in M. vaginatus, act as a tool to organize microbiomes spatially and to attain mutualistic partner specificity in an open, crowded background.
|
|
Scooped by
mhryu@live.com
Today, 1:30 AM
|
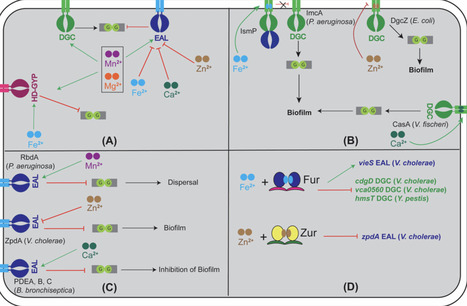
Metals are essential co-factors that are important for the activity of many enzymes and, hence, bacterial life itself. Cyclic di-GMP (c-di-GMP) is an important signaling molecule that is prevalent across various bacterial phyla and involved in myriad physiological processes. The structure and function of diguanylate cyclase (DGCs) and phosphodiesterase (PDEs) domains that make and degrade c-di-GMP are also highly conserved. These enzymes are influenced by several signaling cues, such as temperature, oxygen, and small molecules, which determine the intracellular c-di-GMP levels and subsequent bacterial physiology. In this review, we summarize the current knowledge of the roles of divalent metals Mn2+, Mg2+, Zn2+, Ca2+, and Fe2+ in modulating the activity of DGCs and PDEs and c-di-GMP-related phenotypes. We describe the role of divalent metals in modulating DGC and PDE catalysis, and then discuss the examples of divalent metals as signals that modulate c-di-GMP levels. We also discuss how metals can influence the transcription of c-di-GMP catalytic enzymes. The review highlights the underexplored question of how metal availability shapes c-di-GMP signaling across diverse environmental contexts.
|
|
Scooped by
mhryu@live.com
Today, 1:02 AM
|
The biases revealed in protein sequence alignments have been shown to provide information related to protein structure, stability and function. Recent studies using Potts models show that single-site biases and pair correlations can improve predictions of protein fitness, activity and stability compared to simpler models. Here we use a Potts model to design groups of protein sequences with different amounts of single-site biases and pair correlations and determine the stabilities of sequences from each group. Surprisingly, sequences excluding pair correlations maximize stability compared to sequences that maximize pair correlations, suggesting that pair correlations contribute to other aspects of protein fitness. Consistent with this interpretation, we find that for three enzyme families, activity is greatly increased by maximizing pair correlations. The finding that elimination of covariant residue pairs increases protein stability suggests a route to enhance stability of designed proteins, although this stability may be offset by reduced enzyme activity. Sequence biases at individual positions can be used to design more stable protein variants, while correlation between pairs of residues has also been shown to be important for specifying protein structure and function. Here, a Potts model is used to separate the contributions of single-site biases and pair correlations, revealing that protein structure is stabilized by single-site biases rather than pairwise coupling, while pairwise coupling is important for protein function.
|
|
Scooped by
mhryu@live.com
Today, 12:50 AM
|
Water, the principal component of living organisms including humans, dissociates into H+ and OH- in aqueous environments, and the resulting H+ concentration determines both cellular pH and the proton motive force (PMF) across cellular membranes. These physicochemical parameters are fundamental regulators of a wide range of biological processes. Optogenetics enables the manipulation of biological and cellular functions using light, typically through the ectopic expression of microbial rhodopsins as photoreceptive proteins in target cells or organs. This review provides a comprehensive overview of optogenetic studies employing H+ pump rhodopsins in diverse biological systems and highlights their growing relevance to neuroscience, cell biology, bioengineering, and therapeutic research. Notably, optical control of H+ concentration allows the precise modulation of neural and non-neural activities in animals and bacteria, highlighting the broad applicability and significant potential of H+ pump rhodopsins for optogenetics. Based on previous and current studies, we discuss how further expansion of the molecular toolkit of H+ pump rhodopsins could enable increasingly fine-tuned manipulation of intracellular pH dynamics and PMF for probing cellular physiology and designing next-generation therapeutic strategies, and consider emerging directions that extend beyond classical optogenetics toward the optical control of bioenergetic and chemical processes.
|
|
Scooped by
mhryu@live.com
Today, 12:30 AM
|
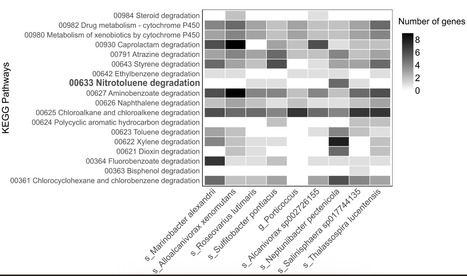
Unexploded ordnance from World Wars I and II continues to release 2,4,6-trinitrotoluene into marine sediments, yet microbial responses to this chronic contamination remain poorly understood. Here, we characterize the taxonomic and functional potential of sediment microbiomes at the historical submarine wreck UC-30 in the North Sea, combining 16S rRNA amplicon sequencing, shotgun metagenomics, and targeted GC-MS/MS analysis with a parallel aerobic laboratory enrichment. Minewell sediments showed distinct community shifts, with enrichment of Proteobacteria, notably Haliaceae and Rhodobacteraceae, alongside increased representation of oxidoreductases and stress-related enzyme classes, including glutathione S-transferases. Genes associated with TNT transformation, including Old Yellow Enzymes and nitroreductases, were modestly enriched in situ. The laboratory enrichment confirmed TNT removal and presence of N-ethylmaleimide reductase, an Old Yellow Enzyme implicated in TNT transformation. Functional and taxonomic parallels between field and enrichment communities indicate shared adaptive capacities under TNT exposure, positioning contaminated marine microbiomes as reservoirs of bioremediation potential. Long-term TNT exposure at historical shipwrecks can influence sediment microbial communities, according to combined wreck sampling from the North Sea and laboratory incubation tests.
|
|
Scooped by
mhryu@live.com
Today, 12:22 AM
|
Accurate enzyme turnover numbers are essential for building enzyme-constrained genome-scale metabolic models. However, collecting and curating these parameters remains a major bottleneck. Indeed, values are scattered across multiple databases, reported under varying experimental conditions, and often missing for many enzymes. To address this challenge, we present WILDkCAT, a Python-based pipeline that enables the retrieval of values from wild-type enzyme measured under user-specified pH and temperature ranges for a given metabolic model. The application to E. coli (iML1515) and Homo sapiens (Human-GEM) models demonstrated the ability of WILDkCAT to retrieve substantial coverage and its applicability across diverse genome-scale models.
|
|
Scooped by
mhryu@live.com
Today, 12:05 AM
|
Microbial communities play fundamental roles in industrial processes and ecosystem stability. However, understanding how individual members and their interactions give rise to community-level function remains challenging because such functions emerge from complex interactions among diverse members. Here, we developed SubCom analysis, a subcommunity-based experimental–computational workflow for inferring candidate taxon-specific contributions and interaction contexts underlying microbial community function. Using an aniline-degrading microbial community, we generated paired composition–function data from 558 randomly assembled, low-complexity subcommunities constructed using a dilution-and-dispense strategy. We then trained decision-tree-based models to predict community function from composition, achieving high predictive performance (r = 0.77–0.89). Interpretation of the learned decision rules identified taxa with consistent functional association: specific Pseudomonas and Acinetobacter taxa were associated with increased community-level aniline utilization, whereas an Achromobacter taxon exhibited a negative association despite its presumed role in downstream metabolism. The models further suggested potential functional interactions, including attenuation of the positive contributions of Pseudomonas and Acinetobacter in the presence of a Corynebacterium taxon, highlighting functional relationships that are not readily inferred from genome-based approaches alone. An augmentation assay using representative isolates supported the predicted direction of several effects and enabled targeted improvement of community function. These results demonstrate the potential of SubCom analysis as a practical framework for inferring taxon-specific contributions and interaction contexts in complex, nonsynthetic microbial communities. Video Abstract
|
|
Scooped by
mhryu@live.com
July 11, 5:03 PM
|
Precise spatial positioning of cellular constituents is critical for bacterial replication, interactions, and behavior. Rod-shaped bacteria may concentrate molecules and machinery at the cell poles, often by leveraging polar scaffolding proteins to recruit other factors. These typically small proteins can self-assemble into higher-order structures that provide platforms to direct fundamental processes like chromosome segregation, division plane selection, and differentiation. Beyond fundamental cellular processes, polarity is implicated in a wide range of bacterial behaviors, including environmental sensing, intercellular interactions, and motility. In this review, we describe polarity-determining factors and their cellular functions specifically in bacterial predators and in host-associated bacteria, including plant and human pathogens. We aim to highlight pivotal roles cell polarity plays in promoting fitness vis-à-vis the host–microbe and microbe–microbe interface.
|
|
Scooped by
mhryu@live.com
July 11, 4:56 PM
|
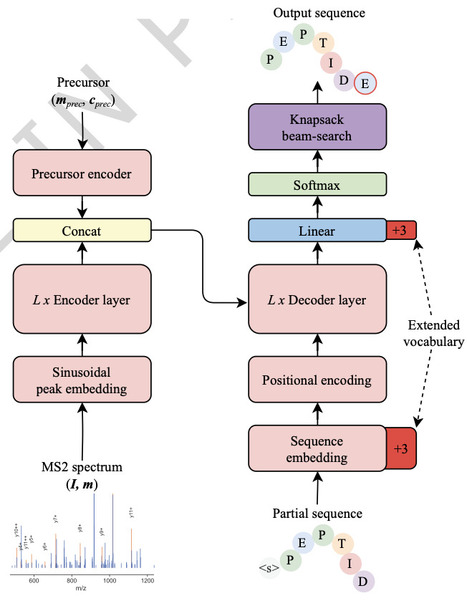
Phosphorylation, a crucial post-translational modification (PTM), plays a central role in cellular signaling and disease mechanisms. Mass spectrometry-based phosphoproteomics is widely used for system-wide characterization of phosphorylation events. However, traditional methods struggle with accurate phosphorylated site localization, complex search spaces, and detecting sequences outside the reference database. Advances in de novo peptide sequencing offer opportunities to address these limitations, but have yet to become integrated and adapted for phosphoproteomics datasets. Here, we present InstaNovo-P, a phosphorylation specific version of our transformer-based InstaNovo model, fine-tuned on extensive phosphoproteomics datasets. InstaNovo-P surpasses existing methods in phosphorylated peptide detection and phosphorylated site localization accuracy across multiple datasets, including complex experimental scenarios. Our model robustly identifies peptides with single and multiple phosphorylated sites, effectively localizing phosphorylation events on serine, threonine, and tyrosine residues. We experimentally validate our model predictions by studying FGFR2 signaling, further demonstrating that InstaNovo-P uncovers phosphorylated sites previously missed by traditional database searches. These predictions align with critical biological processes, confirming the model’s capacity to yield valuable biological insights. InstaNovo-P adds value to phosphoproteomics experiments by effectively identifying biologically relevant phosphorylation events without prior information, providing a powerful analytical tool for the dissection of signaling pathways. Accurate identification and localization of phosphorylation sites remain key challenges in mass spectrometry-based phosphoproteomics. Here, the authors introduce InstaNovo-P, a transformer-based de novo model that enables reliable identification of singly and multiply phosphorylated peptides and uncovers phosphorylation events missed by conventional database-driven approaches.
|
|
Scooped by
mhryu@live.com
July 11, 4:43 PM
|
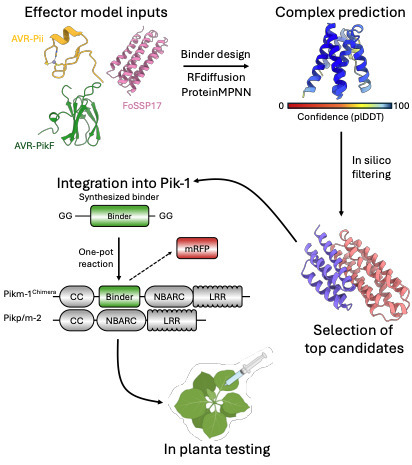
The rapid evolution of plant pathogens poses a persistent threat to global agricultural sustainability, often outpacing the discovery and deployment of natural disease resistance genes. While bioengineering of plant intracellular immune receptors (NLRs) offers a potential solution, developing bespoke immune recognition remains constrained by the laborious characterisation of natural receptors and plant-pathogen interactions. Here, we describe a programmable framework that leverages generative AI protein design tools, RFdiffusion and ProteinMPNN, to design de novo integrated domains (IDs) against diverse pathogen effectors. By integrating these bespoke binders into the modular rice blast Pik-1/Pik-2 NLR receptor chassis, we successfully engineer recognition of a non-cognate virulence factor (effector) from the Panama disease pathogen, Fusarium oxysporum f. sp. cubense Tropical Race 4. Functional assays in Nicotiana benthamiana demonstrate that these de novo domains facilitate specific effector perception and initiate immune signalling, while structural and biophysical analyses confirm that de novo integrated domains maintain high structural fidelity to the initial designs and associate with their targets via the predicted interaction interfaces. Additionally, our findings provide orthogonal evidence for the role of integrated domains in regulation of NLR signalling, demonstrating integration of de novo IDs can either trigger autoactivity or, in some cases, lead to effector-mediated repression of cell death. By decoupling immune perception from natural evolutionary history through deploying AI-designed sensory domains, this work establishes a design-lead framework for generation of programmable plant immune receptors, providing a new avenue for bioengineering crops against emerging pathogens.
|
|
|
Scooped by
mhryu@live.com
Today, 1:39 PM
|
Proteins of unknown function represent a significant gap in our understanding of biological processes, encompassing large portions of the proteomes of many organisms, especially prokaryotes. Addressing this gap is critical to understanding the biology and pathogenicity of such organisms. We introduce ProtPen, an open-source pipeline that facilitates protein function prediction by combining eggNOG-mapper for sequence-based annotation with Foldseek for rapid structural similarity searches using AlphaFold-predicted protein structures. Annotation results from both tools are merged and enriched with UniProt metadata to produce a comprehensive output suitable for downstream analysis. The pipeline requires only a FASTA input file with UniProt identifiers, and is designed to analyze datasets on the scale of whole proteomes. Benchmarking on a curated dataset of well-characterized Pseudomonas aeruginosa proteins demonstrated an annotation accuracy of >90%, and highlighted the complementarity of sequence- and structure-based methods. Further evaluation of ProtPen included its application to biologically relevant datasets, comprising proteins of unknown function that exhibited significant differential abundances in a proteomics dataset of P. aeruginosa, and uncharacterized glycoproteins from Haloferax volcanii. ProtPen is readily extensible to incorporate additional protein function prediction tools. In summary, this pipeline facilitates the systemwide annotation of proteins of unknown function from proteomic datasets and whole proteomes.
|
|
Scooped by
mhryu@live.com
Today, 1:27 PM
|
Large language models (LLMs) are deep learning-based artificial intelligence models that have achieved remarkable success in natural language processing. Typically composed of neural networks with billions of parameters, they are trained on massive unlabeled datasets using self-supervised or semi-supervised learning. Beyond language, LLMs hold immense potential for addressing complex bioinformatics challenges. This review provides a comprehensive overview of transformer-based model applications in genomics, transcriptomics, proteomics, drug discovery, and single-cell analysis. We discuss critical components, including tokenization strategies for diverse biological data, transformer architectures, attention mechanisms, and pretraining approaches. We also survey currently available foundation models and their downstream applications across bioinformatics domains. Finally, we highlight major challenges that remain insufficiently addressed in prior reviews and outline future perspectives and design principles for next-generation biological language models, offering practical guidance for both users and developers.
|
|
Scooped by
mhryu@live.com
Today, 1:20 PM
|
The transition toward a sustainable bioeconomy necessitates the development of robust microbial platforms capable of C1 chemical valorization, particularly the conversion of methanol into high-value chemicals. Komagataella phaffii is a premier methylotrophic host for such applications due to its natural methanol utilization pathways and high-density fermentation capabilities. However, its potential as a C1-based synthetic biology chassis has been hampered by inefficient homologous recombination and the technical complexity of multigene pathway integration. To accelerate the design-build-test-learn (DBTL) cycle for methanol-based biomanufacturing, a more sophisticated and high-throughput genome engineering toolkit is required. We report the development of an HR-enhanced K. phaffii chassis (dKR) achieved by synergistically deleting Ku70 and overexpressing RAD52 at the chromosomal level. This engineering markedly improved the robustness and reproducibility of CRISPR/Cas9-mediated genome editing, increasing integration efficiency from 15.3% in the wild-type to 68.1% in the dKR chassis when using short 50-bp homology arms. The platform demonstrated exceptional capacity for vector-free multi-fragment assembly, achieving an integration efficiency of 86.5% for nine linear DNA fragments. Furthermore, the implementation of a tRNA-ribozyme CRISPR system enabled simultaneous multi-locus integration at three distinct genomic sites with 44.6% efficiency. Notably, the chassis supported direct in vivo plasmid assembly of ten fragments (9 fragments + 1 backbone) with 44.8% efficiency, effectively bypassing E. coli-based cloning. Functional validation via a carotenoid biosynthetic pathway resulted in β-carotene production while maintaining normal cellular growth and metabolic productivity. This study established a versatile genome-engineering platform that advances K. phaffii from a protein-expression host to a broadly applicable biological engineering chassis. By enabling streamlined cellular design and reproducible multigene integration, the platform supports applications spanning synthetic biology, metabolic engineering, and bioproduction. More broadly, the approach illustrates how rational manipulation of DNA repair pathways can enhance genome engineering performance in non-conventional hosts, providing transferable design principles relevant to biological engineering across multiple domains.
|
|
Scooped by
mhryu@live.com
Today, 1:36 AM
|
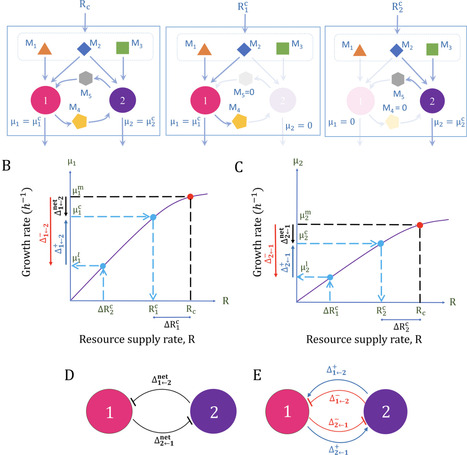
Interspecies interactions are characterized conventionally by the net influence, positive or negative, a species exerts on another. Community ecology theories rely on these net interactions to describe the behavior of multispecies communities. The net interactions in turn comprise positive and negative components, arising typically from cross-feeding metabolites and competition for resources. The components remain challenging to disentangle, compromising descriptions of community behaviour. Here, we devised a method to estimate the components when metabolic interactions predominate. We conceived a theoretical resource partitioning strategy which when applied to data on species growth rates disentangles the components. Consequently, the net influence a species has on another is decomposed into its positive and negative components. The interactions between a pair of species are thus defined by the ‘quartet’ of underlying components, specifically the positive and negative components of the net influence of each species on the other. We applied the method to 28 in silico species pairs from a representative oral microbiome and an experimental auxoptroph pair from the literature. We found that positive and negative components had comparable strengths on average. Interestingly, we found species pairs with similar net interactions but disparate components, highlighting the importance of the quartet. Further, weak net interactions could arise from cancellation of strong components. Estimating the quartet helped better understand the complex transitions in community behavior observed upon varying resource supply in silico and in vitro. The quartet thus offers a more fundamental characterization of interspecies interactions and may help build more reliable community ecology theories, with implications for understanding and design of microbial communities.
|
|
Scooped by
mhryu@live.com
Today, 1:21 AM
|
Plasmid host range (PHR) plays a key role in the spread of ecologically important genes, alongside applications in microbiome engineering and environmental biotechnology. PHR is a complex trait arising from the combination of plasmid, donor and recipient properties. Most studies of PHR use a single donor strain, leaving the role of the donor unexplored and often require genetically tagged recipient strains for counter-selection, which limits the use of non-genetically tractable strains. Here, we applied auxotrophic donor counter-selection in a relatively high-throughput and accessible screening format to characterize PHR across a diverse collection of environmental isolates without the need for recipient engineering. Specifically, we used two auxotrophic donors (Pseudomonas fluorescens and Pseudomonas putida) and plasmid pQBR57-tphKAB, an environmental plasmid engineered for terephthalic acid bioremediation. We screened a library of 101 soil isolates as potential recipients, including genera such as Pseudomonas, Bacillus and Xanthomonas. We only observed conjugation into other Pseudomonas, but donor identity was found to affect PHR, with P. fluorescens conjugating the plasmid into more recipient strains than P. putida. Phylogenomic analysis revealed that transconjugants clustered primarily with the Pseudomonas citronellolis lineage, previously isolated from soil. In strains that were close relatives of transconjugants but unable to acquire the plasmid, we observed five defence systems not present in transconjugants that may act as barriers to plasmid acquisition. Our approach demonstrates how auxotrophic donor counter-selection can be deployed at scale to screen PHR in environmental isolates and to investigate the influence of donor identity on plasmid conjugation.
|
|
Scooped by
mhryu@live.com
Today, 12:52 AM
|
Adenine base editors (ABEs), which enable A•T-to-G•C base editing, have emerged as a powerful tool with potential therapeutic applications. However, conventional ABEs suffer from bystander nucleotide conversions, limiting their utility for precise editing. Here we present a single-nucleotide resolution ABE (snuABE) created by fusing a nickase Cas9, nCas9-H840A, with the deaminase domain of ADAR (adenosine deaminase acting on RNA), which acts on DNA:RNA hybrids, instead of TadA, which acts on single-stranded DNA in conventional ABEs. snuABE requires a target-adenine guide RNA (tagRNA) that introduces a mismatch at the target adenine, enabling highly specific A-to-G editing by ADAR. Engineering ADAR from Pediculus humanus using the in silico protein evolution algorithm EvolvePro, along with 3′-end protection of the tagRNA, enhanced snuABE activity, yielding a median efficiency of 5.4% and a maximum efficiency of 50.0% across 32 targets in HEK293T cells. snuABE exhibits no detectable DNA off-target editing at predicted off-target or orthogonal R-loop sites, highlighting its potential as a precise and safe base-editing technology. Bystander edits are minimized by engineering an adenosine deaminase acting on RNA for DNA base editing.
|
|
Scooped by
mhryu@live.com
Today, 12:47 AM
|
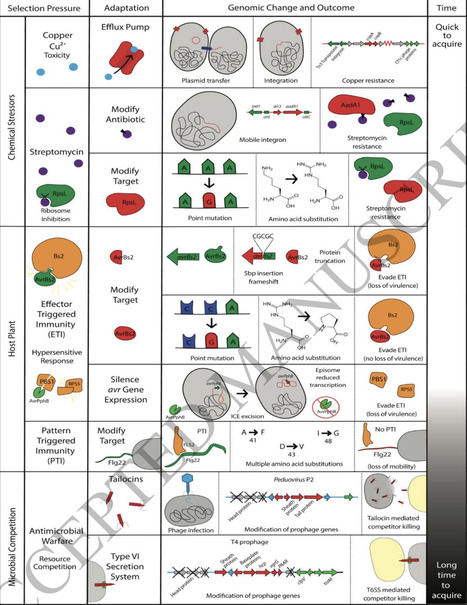
Bacterial plant pathogens have ravaged crops since the dawn of agriculture and continue to pose a serious threat today. Bacteria and their plant hosts have co-evolved in an evolutionary arms race, with artificial selection due to agriculture tipping the scale in favor of the pathogen. This review gives an overview of plant pathogenic bacterial diversity, showing that pathogenicity has independently evolved numerous times, and that there is not one unifying trait determining plant pathogenicity. Instead, these bacteria represent repeated, independent evolutionary transitions driven by life in complex ecological networks, that include plant hosts, insect vectors, microbial competitors, and highly heterogenous abiotic environments. Their genomes reflect this interplay through a dynamic balance of architecture and flux. These structural features, along with highly variable pangenomes, capture the balance between genome stability and flux imposed by ecological constraints and epidemiological dynamics. Horizontal gene transfer via conjugative plasmids, prophages, integrative and conjugative elements, transposons, and in some lineages, natural competence, remains the major source of adaptive novelty, enabling rapid remodeling of virulence repertoires, metabolic capabilities, and antibiotic or heavy metal resistance genes. These changes create distinct selective landscapes. Agricultural practices such as chemical use, host resistance deployment, or seed trade, can drive recurrent bottlenecks, expansions, and admixture events that leave strong genomic signatures in pathogens. Finally, this review explores the genomic differences enabling the divergence of lifestyles, while also acknowledging knowledge gaps and future directions of research on the evolution of bacterial plant pathogens.
|
|
Scooped by
mhryu@live.com
Today, 12:27 AM
|
Plants have evolved two primary innate immune strategies, pattern-triggered immunity and effector-triggered immunity, to prevent excessive microbial infection. As the ‘second genome’ of plants, microbiota also regulates the plant growth-immune tradeoff. In this opinion article, we propose that beneficial microbes expand the plant immune threshold by coordinately regulating above-ground and below-ground immune signaling. We integrate this concept into the existing framework of plant immune strategies to construct a novel model of plant immunity. Building upon this foundation, we introduce a novel perspective regarding dose-dependent immune responses. We propose that, in natural systems, non-pathogen–plant interactions should be evaluated not only based on the specific recognition of microbe-associated molecular patterns (MAMPs) but also by incorporating the dose-dependent effects of MAMPs into the assessment framework.
|
|
Scooped by
mhryu@live.com
Today, 12:09 AM
|
Accurate prediction of peptide structures and peptide–receptor complexes is essential for rational peptide drug development. However, the inherent conformational flexibility of short and disordered peptides presents a fundamental challenge. The AlphaFold model series, which has progressed from AlphaFold2 through AlphaFold-Multimer to AlphaFold3, has substantially advanced computational peptide structure prediction through innovations in geometric reasoning (invariant point attention) and interface-focused confidence metrics (ipTM score), achieving high accuracy for both monomeric peptide structures and multi-chain complexes. However, these models output static conformations, whereas many bioactive peptides adopt their functional conformations only upon binding—often corresponding to low-probability states that static predictions may overlook, leading to failures in virtual screening. This review synthesizes recent advances in the AlphaFold series for peptide studies and applications, discusses their current strengths in structure prediction and receptor-binding analysis, and examines the limitations in capturing conformational dynamics, transient interactions, and chemical modifications. Recent studies have suggested that integrated computational strategies that combine AlphaFold predictions with molecular dynamics simulations, free energy calculations, and ensemble sampling to enhance predictive accuracy and better represent the dynamic nature of peptide–drug interactions. These complementary approaches position AlphaFold as a central computational platform in structure-guided peptide drug design, enabling more efficient lead identification and optimization while bridging the gap between static computational predictions and the complex biophysical reality of peptide therapeutics.
|
|
Scooped by
mhryu@live.com
July 11, 5:06 PM
|
Modern biotechnology depends on a handful of well-characterized microbial species cultivated on sugar-derived feedstocks. Despite their success, these platforms face fundamental constraints: dependence on agricultural resources, vulnerability to contamination, and limited tolerance to unconventional process conditions. Alternative species with native capabilities for one-carbon assimilation, gas fermentation, saline cultivation, or growth at extreme conditions offer compelling solutions, yet their development potential remains largely untapped. Commonly labelled “non-model”, these organisms differ enormously in technological maturity — a distinction critical for assessing feasibility, timelines, and risk. Here, we propose a four-tier engineering readiness framework and apply it to six representative platforms: Moorella thermoacetica, Sporomusa ovata, Xanthobacter sp. SoF1, Methylococcus capsulatus, Halomonas bluephagenesis, and Rhodococcus wratislaviensis. For each, we assess metabolic opportunity space, genetic toolkit development, industrial deployment, and key bottlenecks, illustrating a spectrum from promising wild isolates to tractable engineering platforms.
|
|
Scooped by
mhryu@live.com
July 11, 5:01 PM
|
Virus-like particles (VLPs) are promising for delivering genome editors, yet the in vivo in vivo efficacy of VLP-mediated cytosine base editing remains limited. Here we identified insufficient inhibition of uracil DNA glycosylases as the underlying mechanism of low cytosine base editor (CBE) editing efficiencies in vivo. We engineered a previously reported CBE, transformer base editor (tBE), and developed a VLP delivery system to enhance the recruitment of uracil DNA glycosylase inhibitor proteins. tBE-VLPs achieved robust C-to-T editing in mouse liver and retina. A single injection achieved, on average, 46.0% editing at mPcsk9 and 64.2% at mHpd in the liver, as well as 24.2% at mVegfa in the retinal pigment epithelium, resulting in marked therapeutic benefits in mouse disease models. tBE-VLP4 induced no detectable off-target edits in vitro or in vivo and demonstrated superior specificity compared to AAV or lipid nanoparticle mRNA delivery. Our work establishes tBE-VLP4 as a precise, efficient system for in vivo cytosine base editing. In vivo cytosine base editing is made efficient with potent glycosylase inhibition.
|
|
Scooped by
mhryu@live.com
July 11, 4:50 PM
|
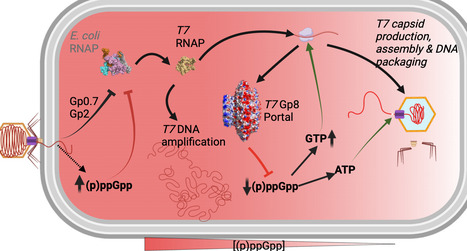
Bacteria restrict viral replication not only through dedicated defense systems but also by entering physiological states that limit cellular resources, yet how phages overcome such host-imposed barriers remains unclear. The stringent response, driven by the alarmone nucleotides ppGpp and pppGpp, can impose a growth-restrictive state that hinders phage infection in specific phage–host contexts. Here, we show that alarmone signaling constrains bacteriophage T7 infection and that the portal protein Gp8 counteracts this barrier by engaging RelA and SpoT, inhibiting their synthetase activities and suppressing alarmone accumulation. Portal mutations that disrupt this interaction sustain alarmone elevation, delay lysis and impair replication in a manner relieved in alarmone-deficient hosts. Portal proteins from representative coliphages share related stringent-response-linked features, indicating that essential virion components can moonlight as antagonists of host stress physiology and that this mechanism is not unique to T7 and may extend to additional coliphages. Bacteria can protect themselves against viral infections by entering physiological states, such as the stringent response, that limit cellular resources. Here, the authors show that phages can use their portal proteins to suppress the bacterial stringent response, thus promoting infection.
|


























system makes use of heterologous (GA) and (GT) attB/attP recognition sequences in the donor vector and the Rosa26 locus, respectively, to deliver the DNA in a directional manner