Your new post is loading...

|
Scooped by
mhryu@live.com
Today, 1:09 AM
|
Reconstitution of functional nitrogenase in plants requires the coordinated expression of the [Fe–S] cluster assembly proteins NifU and NifS. However, the extent to which these proteins interact with endogenous Fe–S metabolism and affect plant physiology remains unclear. Here, we compared NifU and NifS homologs from diverse diazotrophs to identify variants compatible with the plant chloroplast environment. Selected variants of Azotobacter vinelandii, Fischerella thermalis, and Marinobacter lutimaris were characterized by transient expression in Nicotiana benthamiana and stable transformation in rice. Plant-produced NifU was largely devoid of [Fe–S] clusters when isolated but retained strong capacity for in vitro [Fe–S] cluster reconstitution and apo-NifH activation in a Ft > Av >Ml gradient, indicating correct folding and function but limited cluster loading or stability in vivo. NifU and NifS expression in transgenic rice resulted in variant-dependent proteome and phenotype effects, with A. vinelandii-expressing lines exhibiting severe defects, F. thermalis lines showing intermediate phenotype, and M. lutimaris lines being indistinguishable from wild type. These results reveal a trade-off between the biochemical activity of NifU and NifS and their compatibility with host metabolism, which must be considered for successful nitrogenase engineering in plants.
|
|
Scooped by
mhryu@live.com
Today, 12:58 AM
|
The de novo design of ligand-binding proteins has tremendous potential to revolutionize biosensor technology, yet converting these designs into functional sensors remains a major challenge due to the need for ligand-induced conformational changes or modulation of protein-protein interactions. Here, we introduce a physics-based generative approach for the de novo creation of proteins that bind small molecules and metal ions. Our method achieves customizable ligand-binding pocket formation in parallel with simulated protein folding, allowing for precise architectural control of the protein-ligand complex and facilitating the development of biosensors based on either ligand-triggered protein reassociation via split-protein reassembly or ligand-induced protein folding. We demonstrate the versatility of our computational method through successful designs targeting five small molecules, including the very small neurotransmitters serotonin and dopamine, and two metal ions. Biophysical characterization confirmed correct ligand binding, and crystal structures closely matched computational models. We demonstrated the biosensor engineering potential of these designs by constructing serotonin and dopamine sensors using a split protein strategy and explored several approaches to enhance sensor activity. Additionally, we developed a zinc sensor through a zinc-induced protein folding mechanism. Overall, our physics-based generative approach provides a robust framework for the de novo design of ligand-binding proteins, opening new avenues for the development of ligand-responsive biosensors.
|
|
Scooped by
mhryu@live.com
Today, 12:36 AM
|
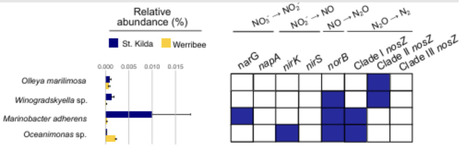
Nearly one-fifth of global emissions of the potent greenhouse gas nitrous oxide (N2O) originate from the ocean, particularly from nutrient-polluted coastal regions. Permeable (sandy) sediments, which cover half of the continental shelf worldwide, are potential sources of N2O due to increasing nutrient inputs from urbanization and agriculture. Yet, the microbial processes determining N2O emissions in these dynamic and unique ecosystems remain understudied. Here, we combined environmental measurements, bacterial cultivation, and genomic analyses to understand the microbes and processes controlling N2O cycling in permeable sediments from Port Phillip Bay (Australia). We established a genomic resource comprising 249 metagenome-assembled genomes and 95 new isolate genomes. Genome-based metabolic reconstructions and culture-based gas measurements revealed diverse bacteria in these sediments produce N2O through incomplete denitrification pathways. However, these bacteria co-occurred with highly abundant clade II N2O-reducing bacteria from the Flavobacteriaceae family. Kinetic profiling showed that both clade II nosZ flavobacterial isolates and whole sand communities exhibited a low apparent affinity for N2O under the tested experimental conditions, expanding the currently limited kinetic data available for N2O reducing microorganisms from coastal permeable sediments, including flavobacterial clade II N2O reducers. Collectively, these findings indicate that abundant N₂O reducing communities can substantially consume N2O within permeable sediments, thus limiting N2O accumulation despite active N2O production. Together with previous hydrodynamic models predicting low N2O release from permeable sediments, our results highlight the important role of specialized microbial communities in regulating N2O cycling under increasing nutrient pollution.
|
|
Scooped by
mhryu@live.com
Today, 12:04 AM
|
Biocontainment systems designed to attenuate the spread of mobile DNA are challenging to evaluate within microbiomes of engineered environments. To better understand how toxin-based biocontainment systems affect horizontal gene transfer (HGT) in a microbiome, we evaluated the host range of pairs of plasmids using orthogonal catalytic RNA (cat-RNA) that amend distinct barcodes to 16S rRNA following HGT. We show that mobilizable (5 kb) and self-mobilizable (60 kb) plasmids, which use the same RP4 transfer machinery but different origins of replication, overlap in their host range when conjugated in parallel into a wastewater community, with 127 of the 143 amplicon sequence variants (ASVs) presenting barcoding signals from both plasmids (89%). We also find that mobilizable plasmids with or without the E. coli CcdB toxin overlap in host range in a wastewater community. Among the two most abundant orders, CcdB attenuated the barcoding signal in Aeromonadales more consistently than Enterobacteriales, which have F plasmids containing the CcdB-CcdA toxin-antitoxin system used for biocontainment. Also, CcdB decreased the abundance of the mobilizable plasmid by >100-fold and yielded mutations in 85% of the reads. Together, these findings reveal how pairs of plasmids expressing orthogonal cat-RNA can be used to monitor the effects of plasmid-encoded traits on mobile DNA persistence following HGT. They also highlight challenges when using biocontainment systems containing genes related to those found in the microbiomes targeted for engineering.
|
|
Scooped by
mhryu@live.com
July 14, 11:34 PM
|
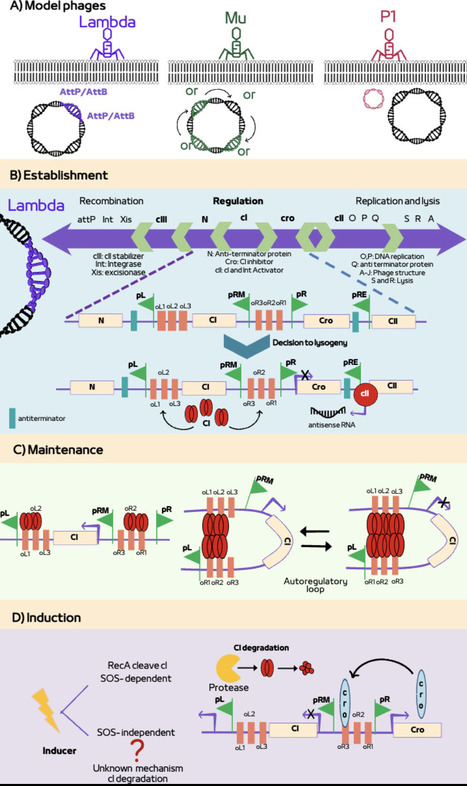
Prophages, as the integrated or plasmid-like forms of temperate bacteriophage genomes, are critical genetic elements that establish stable interactions with their hosts, profoundly impacting host physiology and ecology. Prophages confer multiple advantages to their hosts. They promote changes in the bacterial genome structure and function—such as rearrangements, introduction of novel genes, and gene expression reprogramming—that often enhance bacterial fitness and persistence. Active prophages influence microbial community dynamics through the lytic-lysogenic cycle alternations, while both active and cryptic prophages modulate host behavior, genomic diversification, and interspecific interactions. Given that lysogeny is ubiquitous in nature, prophages play a pivotal role in shaping bacterial ecological functions and evolutionary trajectories. In this review, we provide a comprehensive overview of phage development and the contribution of prophages as modifiers of genome structure and function. We further discuss the long-term consequences of shaping genome architecture in the context of bacterial ecology and evolution, and propose emerging research directions to advance our understanding of phage-host interactions and their impact on microbial ecosystems.
|
|
Scooped by
mhryu@live.com
July 14, 10:33 PM
|
Promoter architecture plays a central role in transcriptional regulation, but predicting promoter activity directly from DNA sequence remains challenging. Here, we tested whether transformer-based DNA language models can learn regulatory logic encoded in Drosophila core promoters. We fine-tuned the pretrained DNA language model DNABERT-2 using a synthetic core promoter dataset measured in S2 cells with luciferase reporter assays. The model predicted promoter activity well when biological replicates were split between training and test data (R² ≈ 0.91), and retained meaningful performance when test promoter sequences were fully excluded from training (R² ≈ 0.64). Model interpretation using SHapley Additive exPlanations (SHAP)1 showed that predictive sequence features matched known promoter elements, including INR, TATA box, DRE, Ohler and MTE/DPE motifs, with position-dependent effects consistent with promoter architecture. Incorporating hormonal activation and nucleosomal context enabled sequence and biological context to be modeled in a unified framework. Gene-wise cross-validation showed promoter-specific generalization across most promoters with promoter-specific differences in accuracy. Applied without retraining to independent Drosophila embryo promoter data, the model captured partial in vivo activity trends. These results show that DNA language models can learn interpretable promoter sequence rules from controlled datasets, while accurate in vivo prediction will require broader regulatory context.
|
|
Scooped by
mhryu@live.com
July 14, 4:48 PM
|
Sequence-to-function models have been very successful in predicting gene expression, chromatin accessibility, and epigenetic marks from DNA sequences alone. However, current state-of-the-art models have a fundamental limitation: they cannot extrapolate beyond the cell types and conditions included in their training dataset. Here, we introduce Corgi, a context-aware sequence-to-function model that overcomes this limitation by integrating DNA sequence and trans-regulator expression to predict chromatin accessibility, histone modifications, and gene expression coverage, even in held-out cell types. Trained on a diverse set of bulk and single-cell sequencing datasets, Corgi achieves top performance in joint cross-sequence and cross-cell-type epigenetic track prediction. Additionally, we present an advanced model version, Corgi+, which is state-of-the-art in imputation of epigenetic tracks using only RNA-seq data. We further show that Corgi learns key cell type-specific trans-regulators in a zero-shot manner, and it can predict genomic variant effects in held-out cell types. This study introduces Corgi and Corgi+ , biology-inspired deep neural network models of regulatory DNA sequences that can generalize to unseen cell types. They accurately impute epigenomic data from RNA-seq experiments, identify key cell type-specific regulators and predict genomic variant effects.
|
|
Scooped by
mhryu@live.com
July 14, 4:36 PM
|
Efficient co-utilization of glucose and xylose is critical for microbial bioconversion of lignocellulosic hydrolysates. However, carbon catabolite repression prevents simultaneous sugar consumption in conventional industrial strains such as E. coli. Here, we blocked the Embden−Meyerhof−Parnas and pentose phosphate pathways in E. coli MG1655 by deleting pgi and gnd, generating strain E. coli MD0 that metabolizes glucose exclusively via the Entner−Doudoroff pathway. Adaptive laboratory evolution yielded mutant E. coli MDE with superior glucose-xylose co-utilization, outperforming E. coli MG1655ΔptsG. Genomic analysis identified gntR and xylR mutations as key contributors to this phenotype. Similar engineering in Klebsiella oxytoca also enhanced glucose and xylose co-utilization. Seven byproduct genes were deleted in E. coli MDE, and efficient production of pyruvate from straw hydrolysate was achieved by using the constructed strain E. coli MDE-6. Further introducing the budRABC operon in E. coli MDE-6 resulted in 2,3-butanediol generation from straw hydrolysate. This study establishes E. coli MDE as a robust chassis for lignocellulose biorefinery.
|
|
Scooped by
mhryu@live.com
July 14, 1:17 PM
|
Discovering functional peptides across vast sequence space remains a formidable challenge, particularly when experimental training data is scarce. We present Minimal Data Maximal Insight (MDMI), a two-stage structure-guided computational pipeline that designs functional peptide variants using only a small, annotated dataset. Rather than relying on sequence information alone, MDMI integrates three-dimensional structural features derived from predicted peptide-protein complexes into a machine learning model that captures interface geometry and binding energetics. This structure-aware predictor, paired with a genetic algorithm for sequence exploration, reduced false positives from 70% to close to zero in an all-negative benchmark panel compared with a sequence-only model in computational benchmarking, and produced approximately four-fold more high-confidence in silico binders than state-of-the-art peptide/protein design baselines. Using the split-GFP system as a testbed, where fluorescence provides a direct functional readout of peptide-protein complementation, MDMI identified peptides with up to 38% sequence divergence from wild-type in Stage 1 while retaining measurable activity. In Stage 2, motif-guided recombination of successful Stage 1 variants produced highly divergent yet functional peptides bearing over 50% sequence difference from wild-type, revealing two distinct functional clusters in sequence space. As further validation, a top-performing candidate expressed as a full-length GFP fusion retained a GFP-like emission profile, supporting formation of a fluorescent GFP-like scaffold. These results demonstrate that structure-informed pipelines can uncover remote functional sequence space from minimal data, with broad implications for peptide and therapeutic analog discovery.
|
|
Scooped by
mhryu@live.com
July 14, 12:10 PM
|
Antimicrobial resistance (AMR) is a major global challenge to human and animal health. The genomic element (e.g. chromosome, plasmid, and genomic islands) and neighboring genes associated with an AMR gene play a major role in its function, regulation, evolution, and propensity to undergo lateral gene transfer. Therefore, characterizing these genomic contexts is vital for effective AMR surveillance, risk assessment, and stewardship. Metagenomic sequencing is widely used to identify AMR genes in microbial communities but fragmentary short-read data do not directly provide this critical contextual information. Assembly of these reads provides some contextual information but fails to recover many mobile genetic elements. Here, we introduce Sarand, a method retaining some of the sensitivity of read-based methods while providing the genomic context of assembly by extracting AMR genes and their associated context directly from metagenomic assembly graphs. Sarand uses BLAST-based homology searches with coverage statistics to identify and visualize AMR gene contexts while filtering false chimeric contexts. Using both real and simulated metagenomic data, we show that Sarand outperforms metagenomic assembly and other recently developed graph-based tools in terms of precision and sensitivity for this problem. Sarand enables effective extraction of metagenomic AMR gene contexts to better characterize AMR evolutionary dynamics within complex microbial communities.
|
|
Scooped by
mhryu@live.com
July 14, 1:40 AM
|
The use of nitrogen (N) fertilizers to meet global food demands is expected to continue rising. However, up to 70% of N applied to agricultural soils is lost through microbially mediated processes such as nitrification. Inhibiting nitrification is thus a key strategy to reduce N losses and improve fertilizer N use efficiency. Various plant-derived compounds, termed biological nitrification inhibitors (BNIs), have been shown to reduce accumulation of nitrification products, intermediates, and byproducts (nitrite, nitrate, nitric and nitrous oxides). However, the mechanisms by which BNIs affect nitrifiers, along with their specificity and persistence in soil are not well understood. Here, we evaluated the effects of three BNIs: methyl 3-(4-hydroxyphenyl) acrylate (MHPA), 6-methoxy-2(3H)-benzoxazolone (MBOA), and limonene, on ammonia-oxidizing, total microbial, and fungal communities in two soils with contrasting pH. Their persistence in each soil was also evaluated. Although ammonia-oxidizing archaea initially dominated nitrifier communities in both soils, their bacterial counterparts significantly increased after mineral N addition but also were more sensitive to BNI application. Limonene and the synthetic inhibitor DMPP stimulated ammonium immobilization, as total soil mineral N was significantly reduced. Limonene and MHPA had the strongest off-target effects, increasing the relative abundance of hydrocarbon-degrading bacteria and potential fungal pathogens, respectively. In contrast, MBOA inhibited nitrification with minimal off-target effects. Among the tested BNIs, MBOA was also the most persistent in the high-pH, high-nitrification-rate soil. Our results show that MBOA is a promising biological inhibitor and highlight the importance of understanding BNIs' ecological effects to develop targeted and sustainable N management strategies.
|
|
Scooped by
mhryu@live.com
July 14, 1:04 AM
|
Novel genes arise through multiple mechanisms, including gene duplication, gene fusion, and horizontal gene transfer (HGT). While HGT has increasingly been documented in animals, the posttransfer evolutionary fate of horizontally acquired genes is less well understood. We hypothesized that fusion with endogenous sequences in animal genomes might generate what we call “HGT-chimeras”: genes with regions of nonmetazoan and metazoan descent in the same open reading frame. To test this hypothesis, we developed a molecular phylogenetics pipeline that enables the identification of HGT-chimeras. We applied our pipeline to 319 high-quality annotated arthropod genomes and uncovered a high-confidence set of 274 HGT-chimeras corresponding to 104 independent origination events across diverse arthropods. HGT-chimeras contain intervals acquired from across the tree of life, and many likely originated via a gene duplication-based mechanism. To assess whether HGT-chimeras might be functionally important, we performed RT-PCR and Sanger sequencing of tissues from 20 arthropod species predicted to harbor HGT-chimeras in their genome. We found evidence for the expression of contiguous chimeric messenger RNA transcripts (mRNAs) for 36 of 41 tested HGT-chimeras across 18 of 20 different tested species. We also found evidence that HGT-chimeras evolve under purifying selection and have acquired potentially functional domain architectures, consistent with the hypothesis that these genes are in active use and may participate in diverse biological processes. These results illuminate an underappreciated combinatorial mechanism underlying the origin of novel genes across the largest animal phylum, and suggest that interdomain sequence fusion can play important roles in animal biology and evolution.
|
|
Scooped by
mhryu@live.com
July 14, 12:27 AM
|
We test the idea that innate agricultural soil microbiomes can be engineered to help support crops under climate change conditions. Salinization is one of the key drivers of agricultural soil degradation globally, and rising sea levels combined with decreasing rainfall are exacerbating this threat to food production. Changes in soil microbial community structures measured from DNA and functions and fitness measured from RNA and labeled amino acid uptakes support the hypothesis that the deliberate use of part-saline irrigation adapts soil microbiomes to increased salinities. While saline irrigation suppressed crop establishment in some cases, this microbiome response combined with inferences of increased microbial nutrient cycling and energy management correlates with final crop yield data and supports the hypothesis that engineered soil communities have helped protect yields under increased salinity conditions. This work provides evidence of the efficacy of a novel pragmatic, cost-effective innate soil microbiome engineering intervention for optimizing and securing food systems for future climate change conditions.
|
|
|
Scooped by
mhryu@live.com
Today, 1:03 AM
|
Biomedical research is increasingly constrained by repetitive, fragmented workflows that slow discovery. We introduce Biomni, a general-purpose biomedical artificial intelligence agent that autonomously executes diverse research tasks. To map the biomedical action space, Biomni’s action-discovery agent mines tools, databases, and protocols from thousands of publications across 25 domains, building a unified agentic environment. Its general-purpose architecture integrates large language model reasoning with retrieval-augmented planning and code-based execution, dynamically composing workflows without predefined templates. Systematic benchmarking shows strong generalization across heterogeneous tasks—causal gene prioritization, drug repurposing, rare-disease diagnosis, microbiome analysis, and molecular cloning—without task-specific tuning. Real-world case studies demonstrate Biomni interpreting multi-modal datasets, optimizing protein stability, orchestrating wet-lab instruments, and generating experimentally testable protocols. Biomni envisions artificial intelligence augmenting human scientists and accelerating discovery.
|
|
Scooped by
mhryu@live.com
Today, 12:53 AM
|
Split aptamer biosensors offer exceptionally low background by assembling only in the presence of a target analyte; however, their performance is frequently limited by the lack of robust design rules for selecting effective split sites. Existing approaches largely rely on heuristic, structure-based assumptions that are poorly validated and often yield suboptimal signal. Herein, we introduce a systematic, data-driven strategy for identifying high-performance split sites within fluorogenic DNA aptamers. Using our massively-parallel aptamer performance analyzer (MAPA) platform, we performed comprehensive single- and double-mutant analysis of the DFAME-binding region of the fluorogenic DNA aptamer Lettuce, informed by its three-dimensional structure. Dimensionality reduction and clustering of the resulting sequence-function landscape revealed mutation-tolerant elements within the binding domain that are suitable for splitting while preserving fluorophore activation. Sensors constructed using these non-intuitive split sites, which are unconventional by standard design principles, exhibited a nearly four-fold improvement in fluorescence signal-to-background ratio for SARS-CoV-2 RNA detection compared to a prior split-Lettuce design. The same split architecture also enabled robust detection of high-pathogenicity H5Nx avian influenza RNA. These results demonstrate that large-scale, data-driven interrogation of aptamer sequence-function relationships can identify non-intuitive split sites and provide a proof-of-concept framework for developing measurement-based design principles for split-aptamer biosensors.
|
|
Scooped by
mhryu@live.com
Today, 12:18 AM
|
DNA has emerged as a promising medium for next-generation information storage due to its ultra-high storage density, long-term stability, and low energy consumption. With the rapid growth of global digital data, DNA storage provides a potential alternative to conventional electronic media. Dynamic random access, which allows selective retrieval of target information without reading the entire dataset, is essential for the practical application of DNA storage. Recent advances in PCR-based indexing, hybridization-assisted retrieval, and electrically controlled addressing have significantly improved access efficiency. However, challenges such as limited primer capacity, amplification bias, and molecular crosstalk still restrict large-scale implementation. This review highlights recent progress, current challenges, and future perspectives for achieving efficient and reliable large-scale DNA data storage systems.
|
|
Scooped by
mhryu@live.com
July 14, 11:59 PM
|
Riboswitches are compact RNA-based regulatory elements capable of modulating gene expression in response to small molecules, without the need for additional proteins. Various synthetic riboswitches have been engineered using in vitro-generated tetracycline and theophylline aptamers. However, many of these constructs exhibit suboptimal switching efficiency and background expression. Moreover, efforts to enhance their performance often involve time-consuming and costly screening processes. Here we report that artificial riboswitches can be efficiently optimized by engineering fusion aptamers that contain two binding pockets (apdimers). Following this rational approach, we generated cooperativity between both binding pockets, resulting in the improved performance of splicing-based and ribozyme-based synthetic riboswitches. We finally combined optimized tetracycline switches, yielding dynamic ranges exceeding 1000-fold with minimal background expression in the OFF state. In addition, we show that the optimized tetracycline riboswitches can be used to efficiently induce AAV-mediated transgene expression in mice. The presented strategy offers a straightforward and effective approach for the optimization of existing synthetic riboswitches and the design of novel riboswitches.
|
|
Scooped by
mhryu@live.com
July 14, 11:30 PM
|
Antimicrobial resistance (AMR) is a global crisis, amplified by slow antibiotic development, inadequate surveillance, and limited understanding of resistance gene dissemination. Traditional detection methodologies fail to capture the complexity and interconnectivity of AMR, highlighting the need for novel approaches. Gut resistome profiling using next-generation sequencing enables untargeted detection of AMR genes within microbial communities, revealing their acquisition, transfer and persistence while providing early warning signals for emerging resistance. This review synthesises advances in resistome science, highlights translational opportunities for infection risk prediction and surveillance, and identifies future directions for integrating gut resistome profiling into precision public health and stewardship frameworks. The gut microbiome contains a vast reservoir of antimicrobial resistance genes. In this Review, the authors discuss how gut resistome profiling is advancing understanding of resistance biology while creating new opportunities for surveillance, clinical risk prediction and antimicrobial stewardship.
|
|
Scooped by
mhryu@live.com
July 14, 5:11 PM
|
Microbial bile acid metabolism is an important link between microbiomes and host physiology, but its genetic basis remains difficult to resolve from genome and metagenome data. This is largely because existing annotation resources are not designed for the high sequence diversity and functional complexity of microbial bile acid genes. Here we present BileActome, a reusable annotation resource developed specifically for microbial bile acid metabolism. BileActome defines 27 experimentally supported gene families, including bile salt hydrolases, bile acid–inducible operon genes, and microbial hydroxysteroid dehydrogenases. Its design prioritizes experimentally supported functional sites when available and conserved domain features otherwise, while also distinguishing key functional subtypes. We applied BileActome to 1,693 high-quality rumen metagenome-assembled and isolate genomes and validated its performance using controlled in vitro rumen fermentations under three bile acid interventions. In metagenomic gene-catalog analyses, BileActome enabled pathway-level interpretation of microbial responses to bile acid exposure, with the most reproducible responses centered on Bai-associated gene families. At genome scale, it generated a phylogeny-informed map of bile acid metabolic potential that was broader and more informative than KEGG-based annotation. Further analyses of genomes, local gene organization, and genome-level guilds showed that bile acid metabolism in the rumen is modular, phylogenetically structured, and distributed across different microbial members. Deconjugation and oxidation/epimerization -related functions were widespread, whereas complete bile acid–inducible systems were less common. Together, these findings support a community-assembled model of bile acid metabolism and establish BileActome as an open and reproducible framework for studying specialized microbial functions in complex ecosystems.
|
|
Scooped by
mhryu@live.com
July 14, 4:38 PM
|
CRISPR–Cas genome editors for fixing faulty genes have been developed over the past few years, but a fresh wave of research is revealing an unexpected way in which related systems can tackle disease: by eliminating the cells that carry faulty genes. Writing in Nature, Scholz et al.1 and Zeng et al.2 show that the enzyme Cas12a2 can be programmed to detect mutations in disease-associated RNA and trigger the destruction of DNA only in cells that express that RNA. The approach could be especially powerful against ‘undruggable’ mutations, including common cancer-associated defects such as those in the gene TP53. Together, the authors’ findings point to a new class of precision therapies that can remove diseased cells while sparing healthy ones.
|
|
Scooped by
mhryu@live.com
July 14, 4:24 PM
|
Synthetic biology has advanced microorganisms to be programmed as production hosts, but its application to bacteria that inherently assemble extracellular materials remains limited. Komagataeibacter spp., natively synthesizes cellulose at the bacterial cell surface, creating a material-forming interface that has not been used as a programmable recruitment platform. Here we establish cell-surface display in Komagataeibacter intermedius and show that this interface can recruit defined proteins, making functionalization part of cellulose formation. By engineering Lpp'OmpA, we displayed a fluorescent protein and genetically encoded capture modules (SpyTag and SilkTag) to selectively capture catcher-fused protein cargos onto K. intermedius cell surface. Recruitment of silk-derived structural protein before cellulose production generated silk-associated fibrous structures within the pellicles, with retained cargo signal after washing. The resulting biocomposite showed reorganized fibre-network morphology, increased surface hydrophobicity, mesoscale ordering, and improved wet-state compressive strength. Wild-type cells exposed to same conditions did not reproduce these changes, demonstrating that material properties arise from surface-directed recruitment rather than protein exposure alone. This work demonstrates the material-forming bacterial surface as a programmable engineering interface for organizing extracellular proteins, providing a general strategy for engineering living materials.
|
|
Scooped by
mhryu@live.com
July 14, 12:35 PM
|
Traits are well-known to recur during evolution, yet how broadly predictable this is at the genetic level remains unresolved. Here, I highlight recent work in plants to underscore that genotype–phenotype repeatability at the level of homologous genes is influenced by mutational effect stability, the extent to which mutations generate consistent phenotypic outcomes across lineages. I propose that observing loss-of-function mutations underlying trait evolution in one lineage increases the likelihood that homologous genes will be reused in convergent evolution across other lineages, whereas observing gain-of-function mutations often indicates reduced predictability at the homolog level. These patterns provide a framework for predicting when evolution will follow homologous genetic routes over long evolutionary timescales.
|
|
Scooped by
mhryu@live.com
July 14, 1:41 AM
|
Spray-induced gene silencing (SIGS) provides a sustainable, highly targeted alternative to chemical pesticides. This review summarizes recent advances in dsRNA technology through a design-to-delivery framework. Key focuses include bioinformatics-driven multi-target dsRNA design for improved stability and efficacy, cost-effective microbial and cell-free synthesis platforms for scalable production, and nanocarriers that protect dsRNA from degradation while enhancing delivery. Integrating rational design, efficient production, and precision nanodelivery will accelerate SIGS from laboratory concept to practical, eco-friendly tool for global crop protection and sustainable agriculture. A review summarizes advances in spray-induced gene silencing (SIGS), covering rational dsRNA design for efficacy, scalable microbial/cell-free production, and nanodelivery systems for stable, effective crop protection.
|
|
Scooped by
mhryu@live.com
July 14, 1:34 AM
|
Biological N2O production from organic nitrogen is generally assumed to require canonical nitrification, which generates oxidized nitrogen that subsequently fuel denitrification. Whether this paradigm universally applies to nitrogen-rich microbial communities remains unclear. Here, we investigated N2O production across an industrial poultry manure composting process and found that substantial N2O formation occurred despite the apparent absence of canonical ammonia oxidation. Neither allylthiourea inhibition nor metagenomic analyses provided evidence for ammonia-oxidizing microorganisms or their activity. Instead, metagenomic analyses identified abundant bacterial nitric oxide synthase (bNos) genes, many of which were phylogenetically affiliated with Bacilli, the dominant bacterial group throughout composting. Physiological experiments with Bacillus isolates demonstrated a nitrification-independent route in which L-arginine was oxidized to NO2⁻/NO3⁻, consistent with bNOS-mediated NO formation followed by abiotic oxidation. Recovery of 15N-labelled N2O following 15NO2⁻ addition established NO2⁻ as an immediate precursor of aerobically produced N2O, confirming that the oxidized nitrogen generated through this alternative route subsequently fueled denitrification. Metagenomic analyses further revealed extensive denitrification potential but comparatively low nosZ abundance. Together, these findings identify a previously overlooked route linking organic nitrogen turnover to denitrification independently of canonical nitrification, thereby expanding current models of microbial N2O production in composts and potentially other protein-rich thermophilic environments.
|
|
Scooped by
mhryu@live.com
July 14, 12:43 AM
|
Cell division is a fundamental process essential for life, underpinning reproduction, development and tissue maintenance across all organisms and enabling population growth and evolutionary adaptation. Recreating this capability is, therefore, a central challenge in bottom–up synthetic biology, wherein the aim is to construct functional synthetic cells. In recent years, substantial progress has been made toward building a synthetic divisome through partial reconstitution of the protein machinery underlying cell division in vitro. Here, we review current strategies to mimic the key stages of division: symmetry breaking to define the division site, membrane deformation to drive constriction and, thus, shape changes of the cell, and the final abscission event. We critically assess the successes and limitations of these approaches and discuss how integrating multiple modules may enable the realization of a minimal, functional division system for synthetic cells. Cell division consists of three elementary steps: symmetry breaking, membrane constriction and abscission. Here, we review current strategies for achieving these steps in synthetic cells, assessing their successes and limitations with a view to integrating them within a divisome for synthetic cells.
|