 Your new post is loading...

|
Scooped by
mhryu@live.com
Today, 5:04 PM
|
|
|
Scooped by
mhryu@live.com
Today, 4:38 PM
|
The Type VI CRISPR-Cas13 system, evolved from prokaryotic immunity, has become a versatile, programmable RNA-targeting platform with broad biotechnological potential. Guided by CRISPR RNA (crRNA), Cas13 cleaves single-stranded RNA via higher eukaryotic and prokaryotic nucleotide-binding domains, exerting specific (cis) and nonspecific (trans) collateral cleavage, which enables ultrasensitive nucleic acid detection while introducing cytotoxicity risks in eukaryotic cells. Diversification of Cas13 subtypes, including compact variants, enhances targetability and delivery compatibility, and inhibitory strategies (anti-CRISPR proteins, crRNA mimicry/degradation) enable activity modulation for improved safety. Building on mechanistic foundations, Cas13 is repurposed for targeted RNA knockdown, nucleic acid diagnostics, live-cell RNA imaging with catalytically inactive variants, programmable RNA base editing through deaminase fusions, splicing regulation, epitranscriptomic editing of multiple RNA chemical marks, interaction mapping of RNA–protein and RNA–RNA networks, and translational control, with preliminary clinical translation in antiviral therapies, pathogenic transcript correction, and cancer therapy. Furthermore, Cas13-integrated diagnostics and functional genomics are accelerating biomarker discovery and personalized treatment. Nevertheless, successful clinical translation hinges on overcoming critical bottlenecks, including tissue-specific delivery, mitigation of collateral cytotoxicity, and management of host immunogenicity. This review synthesizes Cas13 classification, structure–function principles, regulatory inhibitors, application modalities, and translational challenges to inform next-generation engineering and responsible deployment of RNA-targeted technologies.
|
|
Scooped by
mhryu@live.com
Today, 4:22 PM
|
Natural biological CO2 fixation converts atmospheric CO2 to energy-dense carbohydrates, thereby providing an alternative to fossil fuels and contributing to the restoration of carbon balance. Recent advancements in the mechanisms of biological CO2 fixation have led to innovative architectures of CO2 fixation pathways and energy systems that exceed the efficiency of natural carbon assimilation. Synthetic CO2 bioconversion systems (SCBS) have been designed and reprogrammed for the carbon-neutral or carbon-negative biomanufacturing of chemicals from CO2 by integrating multi-carbon biosynthesis with various energy conversion methods, including light, electrical, and chemical processes. In this review, we systematically analyze how to achieve efficient matching of CO2 fixation modules with energy supply modules, aiming to establish scalable SCBS for addressing the pressing issue of atmospheric CO2 overload. Then, we propose a systematic framework for designing next-generation biomanufacturing with enzymatic or microbial CO2 bioconversion systems, facilitating the construction and optimization of SCBS towards carbon-neutral or carbon-negative bioproduction. Furthermore, we emphasize transformative SCBS technologies, such as photo-biohybrid systems for converting light into chemical energy, electro-biohybrid systems for transducing electrical energy into chemical forms, and enzyme cascade systems for repurposing chemical energy, all of which aim to achieve unprecedented efficiency in powering biosynthesis from CO2. Finally, we propose a strategic roadmap for carbon-negative biomanufacturing ecosystems, wherein bioinspired CO2 fixation platforms can synergistically integrate with the principles of a circular economy to facilitate the industrial transition to net-zero emissions.
|
|
Scooped by
mhryu@live.com
Today, 4:06 PM
|
Traditional proximity biotinylation approaches require extensive genetic engineering or intricate purification steps. Here, we introduce FISH+, a ready-to-use RNA proximity labeling method that relies on the recruitment of peroxidase to RNA targets, and facilitates in situ proximity biotinylation in fixed cells. This method permits concurrent visualization of RNA molecules and identification of RNA-interacting proteins. Using this method, we visualized 45S and NEAT1 RNA, and captured their proximal proteins, demonstrating the capability to concurrently visualize RNA and identify proximal proteins. By targeting PNCTR (~36 copies per cell), we observed distinctly bright RNA dots, representing the combined biotinylation signals from both RNA and proximal proteins in situ. This indicates the potential of the FISH+ method for enhanced RNA visualization. We further generalized the FISH+ method to explore XIST-interacting proteins, a number of reported interactors were significantly enriched, such as SPEN, CIZ1 and RBM15. Using quantitative mass spectrometry, we show that FISH+ correctly identifies known RNA-protein interactions in the nucleus of human cells. Overall, we established a watch-and-catch punctate RNA method through the integration of RNA fluorescence in situ hybridization (FISH) with proximity biotinylation. This method provides additional spatial information for the characterization of RNA-centric interactions in fixed, genetically unperturbed samples. FISH+ extends RNA FISH from visualization to protein identification through a simple, robust and highly accessible “watch-and-catch” proximity labeling method for punctate RNAs.
|
|
Scooped by
mhryu@live.com
Today, 3:54 PM
|
Biomolecular condensates are emerging structures that organize cell biochemistry. RNA-protein (RNP) condensates have raised huge interest in the field of RNA biology due to their potential to impact gene expression. Although RNP condensate biophysical properties and assembly mechanisms have been extensively studied, leading to major breakthroughs, their contribution to biological processes remains debated. In this perspective, we review the current knowledge on the functions of cytoplasmic RNP condensates in mRNA regulation. Particularly, we highlight recent technological and conceptual advances that revealed the unexpected function of RNP condensates in mRNA translation. We discuss the mechanisms and biophysical bases that reconcile RNP condensate dual function in translational repression and activation. We propose emerging future directions to further address translation at RNP condensates and decode their functional compartmentalization linked to their biophysical properties. We also highlight the importance of this new function of condensates in translation for improved RNA-based therapeutics.
|
|
Scooped by
mhryu@live.com
Today, 1:00 AM
|
The bacterial 16S rRNA gene is widely used to characterize host-associated and environmental microbiomes, most commonly through sequencing short hypervariable regions. Recent improvements in PacBio sequencing chemistry and concatenation approaches can now enable high-throughput, full-length 16S rRNA gene sequencing with high accuracy and depth. However, errors introduced during library preparation remain a major limitation, particularly during PCR amplification of full-length amplicons, where error accumulation may be elevated due to longer sequence lengths. These challenges are amplified when samples vary widely in microbial biomass, making it difficult to select a single optimal number of PCR cycles. Here, we evaluated PCR cycle autonormalization for PacBio Kinnex full-length 16S rRNA gene sequencing across seven agriculturally relevant specimen types. We compared conventional fixed-cycle PCR protocols (20, 24, and 30 cycles) with an autonormalization approach in which individual reactions were terminated during exponential amplification based on real-time fluorescence thresholds. Under the workflow tested here, autonormalized libraries generally retained a high proportion of sequences following denoising and chimera removal, exhibited low residual error rates (<0.005%), and yielded relatively even read distributions across heterogeneous sample inputs. Overamplified reactions (30 cycles) showed elevated residual error rates and greater sequence loss, particularly in samples with higher microbial biodiversity, whereas low-cycle libraries produced more variable read output among specimens. Importantly, the PCR protocol had relatively minor effects on overall community composition compared with specimen type. These results support PCR cycle autonormalization as a useful workflow strategy for heterogeneous full-length 16S library preparation, while also highlighting the importance of library design, pooling strategy, and downstream processing in shaping technical outcomes.
|
|
Scooped by
mhryu@live.com
Today, 12:42 AM
|
RNA polymerase catalyzes transcription, the first step of gene expression. In bacteria, numerous regulatory proteins and signaling molecules fine-tune RNAP activity in a promoter-specific manner. The resultant changes in gene expression allow cells to acclimate to an ever-changing environment. In addition to phenotypic adaptation, increases in cell fitness can also result from changes in the genome. Here, we explore how mutations in RNAP structural genes benefit cells under diverse selection pressures, with a focus on antibiotics. Selection for resistance to rifampicin (RIF), an antibiotic that binds near the catalytic center of RNAP, leads almost exclusively to amino acid substitutions in the large β subunit that modify the RIF binding site. RIFR mutations have pleiotropic effects and can lead to increased or decreased sensitivity to other antibiotics. In addition, mutations in RNAP are linked to resistance to β-lactams, antibiotics that target peptidoglycan synthesis. Mutations in RNAP can act by altering the interaction with key regulators, including the sigma (σ) factors required for promoter recognition, transcription factors, or signaling molecules that bind to RNAP. RNAP mutations also affect catalysis with impacts on promoter recognition and clearance, elongation, and termination. We consider illustrative examples of changes in RNAP that alter the transcriptional landscape to facilitate the emergence of antibiotic tolerance and resistance, both in the laboratory and during the clinical course of treatment in patients.
|
|
Scooped by
mhryu@live.com
June 22, 11:47 PM
|
The twin-arginine translocation (Tat) system is a mechanistically unique protein transport pathway moving folded proteins across membranes. It is found in all domains of life and is essential for bacterial virulence and plant photosynthesis. The membrane proteins, TatA, TatB and TatC form a core complex to which substrate proteins bind, triggering the recruitment of additional TatA protomers to form the transport site. Here we present cryo-electron microscopy structures of the prototypical TatBC complex from E. coli and the atypical complexes from Nitratifactor salsuginis and Myxococcus xanthus in a resting state, alongside TatAC substrate-bound TatBC and TatABC complexes from E. coli in the early stages of transport. These structures demonstrate that substrate proteins associate with the core complex solely through their N-terminal signal peptides. The Tat targeting sequences of the signal peptides make specific contacts with TatC, and the peptide body is clamped by TatB. The core complex contains highly tilted transmembrane helices that drive extreme local membrane thinning. On the basis of our structures and biochemical and functional analyses, we propose a model for the early steps in Tat transport. Cryo-EM structural, biochemical and functional analyses of the bacterial twin-arginine translocation system for protein transport across membranes reveal mechanisms of substrate binding.
|
|
Scooped by
mhryu@live.com
June 22, 11:28 PM
|
Bacterial consortia, with broad metabolism and environmental resilience, show promise for bioaugmentation treatment of plastic wastes. Consortia design principles and plastic degradation enhancements in laboratory and simulated-system studies were reviewed. Practical barriers include narrow polymer scope, long treatment duration, limited scalability, ecological risks, and scarce techno-economic assessments. Bioaugmentation can be realized through the development of bacterial formulations, the integration of pretreatment–bioaugmentation workflows, and the implementation of long-term field trials.
|
|
Scooped by
mhryu@live.com
June 22, 11:23 PM
|
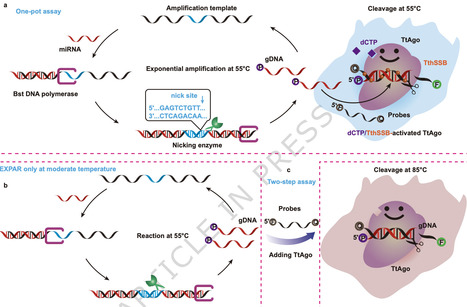
Thermus thermophilus Argonaute (TtAgo) is a DNA-guided endonuclease promising for gene editing and molecular diagnostics. However, its high-temperature dependence (65–85 °C) restricts its utility in moderate-temperature scenarios. Here we show that combining deoxycytidine triphosphate (dCTP) with the single-stranded DNA-binding protein from T. thermophilus (TthSSB) enables robust TtAgo activity within 37–60 °C. Mechanistic studies reveal that dCTP increases TtAgo’s conformational flexibility, while TthSSB promotes substrate recruitment, synergistically driving efficient DNA cleavage. Leveraging this mechanism, we develop ERCB-TtAgo (exponential amplification reaction by dCTP/TthSSB-activated TtAgo), a one-step isothermal diagnostic platform for microRNA detection. It achieves femtomolar sensitivity within 40 min and accurately distinguishes hepatocellular carcinoma patients from controls in a 151-sample clinical cohort, matching RT‑qPCR performance. Our work not only surmounts a key limitation of TtAgo but also offers an approach for modulating nuclease activity, paving the way for intelligent biosensing platforms with broad applicability. Thermus thermophilus Argonaute (TtAgo) requires high temperatures for activity. Here, authors show that dCTP and a single stranded DNA binding protein enable TtAgo to work at moderate temperatures, allowing them to develop an isothermal miRNA detection platform for the diagnosis of hepatocellular carcinoma.
|
|
Scooped by
mhryu@live.com
June 22, 10:37 PM
|
Ammonia-oxidizing archaea (AOA) are widespread and highly abundant in nature. Despite their typical aerobic metabolism, they thrive in ecosystems where oxygen is scarce. Recently, the AOA Nitrosopumilus maritimus SCM1 was shown to produce oxygen and N2O from nitrite upon oxygen depletion, with N2O subsequently converted to dinitrogen (N2) supporting nitric oxide (NO) dismutation as the proposed metabolic pathway. Here, we explore the ability of other ammonia oxidizers, with diverse phylogenetic affiliations and from different environmental settings to produce oxygen via NO-dismutation. We studied three marine AOA, one soil AOA and two soil ammonia-oxidizing bacteria (AOB). Upon oxygen depletion, all strains accumulated oxygen. In incubations of the AOA strains with 15N tracers, two of them, Nitrosopumilus piranensis D3C and Nitrosopumilus adriaticus CCS1, showed transient 46N2O accumulation followed by linear 30N2 production, similar to SCM1, while Nitrosopumilus adriaticus NF5 and Nitrososphaera viennensis EN76 mainly produced 46N2O from nitrite without further N2 accumulation. These findings indicate that oxygen production through NO-dismutation is a common metabolism in cultured representatives of AOA, albeit with physiological variations between different strains such as the reduction of N2O to N2. Comparative genomics did not reveal a distinct putative N₂O reductase in the AOA strains with linear N2 production, suggesting that the observed physiological differences may not directly stem from gene inventory. The finding of oxygen production in several AOA, as well as AOB, suggests that dark oxygen production is a common trait among archaeal and bacterial nitrifiers and adds a potential explanation for their abundance in oxygen-depleted environments.
|
|
Scooped by
mhryu@live.com
June 22, 10:26 PM
|
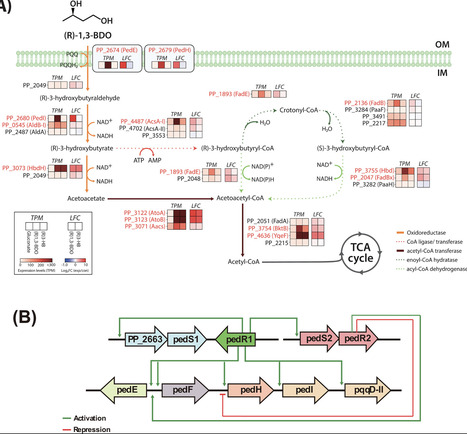
1,3-Butanediol (1,3-BDO) is widely used in consumer and industrial products; however, its microbial degradation remains poorly understood. Here, we dissect the catabolic and regulatory mechanisms of (R)−1,3-BDO utilization in Pseudomonas putida KT2440 and develop (R)−1,3-BDO-responsive transcriptional biosensors. Transcriptomics and qRT-PCR revealed strong induction of the ped gene cluster, which oxidizes (R)−1,3-BDO to (R)−3-hydroxybutyrate [(R)−3-HB], and of the LysR-regulated operon PP_2047–2051, which channels (R)−3-HB toward acetoacetate and acetyl-CoA. Gene deletion and enzyme assays identified pedE and PP_2049 as essential for (R)−1,3-BDO catabolism, with PP_2049 playing a more critical role than the canonical β-hydroxybutyrate dehydrogenase HbdH (PP_3073). Regulatory analysis showed that PedR1 directly activates catabolic genes independently of PedR2—challenging the widely accepted indirect-only model—while PedS1 proved dispensable, implying an alternative sensor kinase. Promoter–GFP fusions demonstrated that full activation of the pedE promoter requires both PedR1 and the PedS2–PedR2 two-component system, whereas the pedS2R2 promoter requires only PedR1 and functions in both native and heterologous hosts, including E coli. These results define the genetic and regulatory architecture of (R)−1,3-BDO degradation in P. putida and establish pedE- and pedS2R2-based promoters as (R)−1,3-BDO-responsive biosensors.
|
|
Scooped by
mhryu@live.com
June 22, 7:29 PM
|
Precise and efficient replacement of large genomic DNA segments without inducing double-strand breaks (DSBs) remains a central challenge in genome engineering. Traditional homologous recombination relies on DSBs and long homologous arms, yet it remains inefficient, while recombinase or integrase systems suffer from residual sequences at integration sites. Prime editing (PE), limited by the processivity of reverse transcriptase, struggles to integrate large fragments (>100 bp). To address this challenge, we introduce Prime Editing–Microhomology-Enabled Replacement (PREMIER), a DSB-free platform by installing single-stranded microhomology arms at donor and genomic junctions via PE. In cell lines, PREMIER achieved a mean efficiency of 63.4% (median 65.2%) in diverse target sites, with peak efficiencies reaching 85.9%, exceeding homology-directed repair by 10–20-fold and reducing off-target integrations by over 100-fold compared to nonhomologous end joining. It bypasses the need for long homology arms, simplifies donor preparation, achieves targeted replacement of sequences up to 10.3 kb. In vivo, PREMIER integrates a 6.2-kb oncogene cassette into the mouse liver. Additionally, PREMIER replaces murine Trp53 with human TP53 CDS, generating functional humanized mice. Altogether, PREMIER provides a precise, high-efficiency, and DSB-free strategy for large-scale genome rewriting, offering a powerful tool for complex modeling and therapeutic genome editing.
|
|
|
Scooped by
mhryu@live.com
Today, 5:01 PM
|
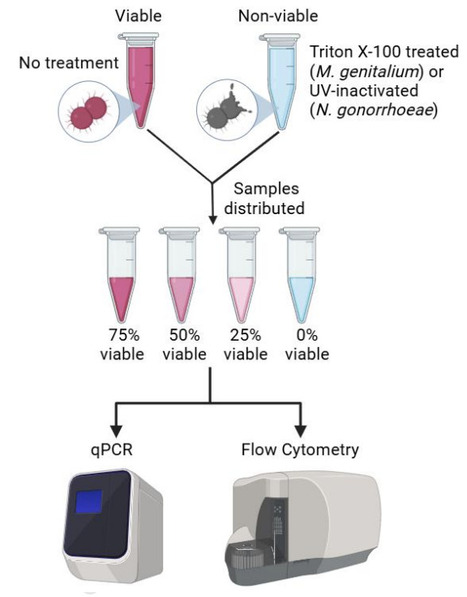
Neisseria gonorrhoeae and Mycoplasma genitalium infect more than 150 million people each year and are rapidly acquiring antimicrobial resistance (AMR), to all available treatments. PCR tools used for diagnosis and test-of-cure, amplify nucleic acids regardless of microbial viability, leading to false positives and subsequent overuse of antibiotics. Assays that accurately assess microbial viability and replication after exposure to antibiotics and other stimuli are needed to improve diagnostics. In this study, we present a flow cytometric method to quantitatively assess N. gonorrhoeae and M. genitalium viability and replication. We utilised a membrane exclusion dye (Fixable Viability Stain 700) to differentiate live and dead cells and a cell trace dye (CellTrace Violet) to track cell replication. The flow cytometry assay was assessed with and without nutrient starvation and inactivation (permeabilisation or UV exposure) to validate the assays' ability to resolve replicating, non-replicating, and non-viable bacterial populations. The outcomes of flow cytometry were compared to qPCR assays and culture-based approaches. Whereas qPCR overestimated viable cell counts (two-to-four-fold overestimation) and could not distinguish between viable and non-viable cells (p > 0.05), flow cytometry could reliably distinguish these populations, as well as replicating and non-replicating subpopulations. This novel flow cytometry method can be used to improve upon existing techniques for in vitro experimentation such as antimicrobial sensitivity testing and could be further utilised for cell sorting, enabling downstream analyses at the single-cell and population level, providing a basis for the discovery of viability targets for diagnostic tools.
|
|
Scooped by
mhryu@live.com
Today, 4:28 PM
|
Nanobody–antigen molecular recognition underpins nanobody discovery and development, necessitating accurate determination of binding occurrence, interface residues, and affinity. Current predictors are architecturally designed for the massive, heterogeneous spectrum of general protein–protein interactions, diluting the limited, complementarity-determining region (CDR)-dominated nanobody–antigen interaction (NAI) data and masking the decisive CDR signal. The scarcity of experimental affinity data precludes direct regression-based estimation of binding affinity. Here, we present NanoBind, a mechanism-driven deep learning framework that embeds the CDR-dominated binding pattern within its encoder, enabling robust prediction of binding occurrence and interface residues from limited NAI data. Constrained by scarce affinity data, NanoBind generates quantitative affinity ranges for nanobody–antigen pairs without extra experiments. Systematic benchmarking demonstrates that NanoBind surpasses state-of-the-art methods in accuracy and robustness, and interpretability analyses confirm that the model’s decisions align with the CDR-dominated binding mechanism. When million-sequence immune repertoires are screened against 4 antigens, NanoBind reduces candidate nanobodies to fewer than 100 per target. For the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) spike receptor-binding domain (RBD)–nanobody F2 complex, NanoBind correctly predicts binding occurrence, matches experimentally validated interface residues, and generates an affinity range quantitatively supported by molecular dynamics simulations. A server is available at http://liulab.top/NanoBind/server.
|
|
Scooped by
mhryu@live.com
Today, 4:15 PM
|
Machine learning (ML) has the potential to revolutionize antibody design and selection, but its success depends on access to well-curated datasets of antibody-antigen interactions. We developed a synthetic Fab yeast display library optimized for seamless integration with ML processes, focusing on sequence diversity within the complementary determining region heavy chain CDRH3 loop. The library incorporates key sequence features derived from human B cell repertoires captured in a compact antigen recognition module (ARM) format. Built with the VH1-69 heavy chain and four light chains, the library was evaluated against ten human and murine cell surface antigens, including programmed cell death ligand 1 (PD-L1), T cell immunoreceptor with immunoglobulin and immunoreceptor tyrosine-based inhibitory motif domains (TIGIT), and roundabout guidance receptor 1 (ROBO1). This approach yielded hundreds of antibodies with robust biophysical properties, some of which were validated by flow cytometry and immunohistochemistry. Furthermore, ML analysis identified additional antibodies for ROBO2 and PD-L2 from the aggregate sequencing data. The publicly available dataset establishes an ML-compatible framework designed to accelerate and streamline antibody discovery and development.
|
|
Scooped by
mhryu@live.com
Today, 3:58 PM
|
The fermented food microbiome comprises live microorganisms, their genetic elements and their metabolites, and represents an established dietary approach for modulating host–microbiome interactions through the consumption of fermented foods. Fermentation enhances food preservation and nutrient bioavailability, and supplies the host with probiotics, prebiotic substrates and postbiotic metabolites. These bioactive compounds can influence the oral and gut microbiota, modulate immune function and support metabolic resilience. Fibre-rich, plant-based fermented foods retain such components within structured matrices that enhance microbial viability and mucosal interactions more consistently than do fermented dairy foods. This Review explores how the fermented food microbiome affects the oral–gut axis via both transient microbial exposure and metabolite-mediated signalling. Drawing on clinical and preclinical evidence, we examine how fermented food intake alters resident microbiota and host physiology throughout the digestive tract. Despite growing evidence, the mechanisms through which fermented food might promote health remain insufficiently defined in humans owing to strain variability, inconsistency in microbial composition across fermented foods, heterogeneous clinical outcomes and regulatory ambiguity. Taking into account these limitations, we propose a roadmap to integrate the fermented food microbiome into precision nutrition as a feasible, personalized, diet-based strategy to promote health and prevent disease. The bioactive compounds and microbial consortia contained in plant-based fermented foods support oral and gut microbiota, host immune function and metabolic resilience. This Review describes the impact of the fermented food microbiome on the oral–gut axis and identifies a strategic roadmap to integrate this edible microbiome into precision nutrition practices to promote health.
|
|
Scooped by
mhryu@live.com
Today, 3:33 PM
|
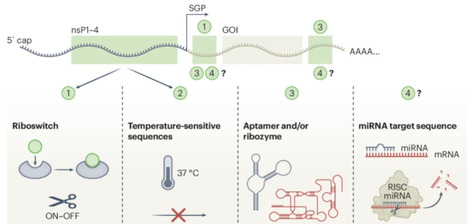
Precise, reversible control of gene expression from self‑amplifying RNA (saRNA) remains difficult, limiting the therapeutic flexibility of this otherwise potent platform. Although alphavirus‑derived saRNAs encode non‑structural proteins that drive RNA replication and offer an intrinsic regulatory point, no existing approach has enabled direct, drug‑dependent control of this machinery for high‑fidelity modulation of expression. Here we engineer saRNA constructs whose replication is activated by the approved small‑molecule drug trimethoprim, using drug‑responsive degradation domains fused to individual non‑structural proteins to regulate self‑amplification. As each replication protein contributes differently to RNA copying, we systematically screened fusion configurations and identified an optimal design combining modified replication proteins with a regulated payload. This construct achieved more than a 104‑fold difference between on and off states with negligible background expression. In mice, oral trimethoprim enabled tunable, reversible and temporally programmed expression patterns. When encoding a human immunodeficiency virus antigen, an escalating trimethoprim regimen enhanced germinal centre responses, a key determinant of antibody affinity maturation. This drug‑regulated saRNA platform provides a controllable and clinically compatible strategy for vaccines, immunotherapies and gene therapies. A self-amplifying RNA circuit whose replication is activated by an FDA-approved small molecule enables precise, reversible and programmable control of RNA amplification and antigen expression for improved therapeutic and vaccine design.
|
|
Scooped by
mhryu@live.com
Today, 12:52 AM
|
The engineered Photorhabdus virulence cassette (PVC) enables precise protein delivery but has not yet achieved RNA packaging and injection delivery. In this study, we achieved intraluminal RNA loading via the U1A RNA-binding domain, anchoring it to the PVC inner tube and establishing DART (PVC Docker-based All-purpose RNA Injection Delivery Tool). This enabled the protective loading of diverse RNAs, including Pepper RNA, guide RNA, siRNA, miRNA, and mRNA. Through the co-delivery of Cas9 in vitro, DART also drove effective knockouts of enhanced green fluorescent protein gene (EGFP), Kirsten rat sarcoma viral oncogene homolog (KRAS), and programmed death-ligand 1 (PD-L1), representing reporter, oncogenic, and immune-related targets for evaluating DART-mediated gene editing. Notably, DART-mediated KRAS knockout produced a significant antitumor effect in a subcutaneous mouse tumor model. Complementary to the external spike-surface fusion strategy of SPEAR (a PVC system termed spike engineering and retargeting), as an intraluminal nanosyringe platform, DART employs internal engineering to expand PVC from protein to RNA delivery.
|
|
Scooped by
mhryu@live.com
Today, 12:38 AM
|
The microbiome actively influences antimicrobial resistance (AMR) dynamics by shaping both ecological and evolutionary processes. However, the extent of its role in resistance emergence, transmission and persistence remains unclear. Traditional AMR research has mainly focused on genetic mechanisms and pathogen-level dynamics. In contrast, the intersection of AMR and the microbiome, including resistance-gene reservoirs, microbial competition and community-mediated selection, remains poorly represented, especially in a modelling context. Here we present a structured framework for incorporating microbiome–AMR interactions into predictive models. We identify key microbiome-mediated processes shaping AMR across different levels of complexity, describe how these can be quantitatively integrated into models, and identify critical data gaps that limit current approaches. By bridging microbiome ecology, AMR biology and mathematical modelling, we set out research priorities and strategies to improve resistance prediction and guide microbiome-targeted interventions. The microbiome plays a significant yet underexplored role in antimicrobial resistance by influencing ecological and evolutionary processes. This Perspective proposes a framework to integrate microbiome–AMR interactions into predictive models while highlighting key mechanisms and data gaps to improve resistance understanding and interventions.
|
|
Scooped by
mhryu@live.com
June 22, 11:37 PM
|
Microbial inoculants are increasingly promoted as sustainable alternatives or complements to conventional agricultural inputs, yet their field performance remains highly variable. This review examines how ecological processes governing root microbiome assembly constrain inoculant establishment, persistence, and function across agricultural systems. We synthesize current evidence on the roles of environmental filtering, host-mediated selection, microbial interactions, and context dependency in shaping inoculant outcomes. We further evaluate the promise and limitations of core-microbiome concepts and synthetic communities as emerging strategies for microbiome-informed inoculant design, emphasizing that their practical translation remains challenged by methodological variability, ecological complexity, formulation constraints, and regulatory barriers. By integrating ecological theory with applied microbiology, this review highlights why many inoculants fail to deliver consistent agronomic benefits and outlines a more potential framework for developing context-aware, field-relevant microbiome-based solutions for sustainable agroecosystem management.
|
|
Scooped by
mhryu@live.com
June 22, 11:25 PM
|
Plastic pollution poses a severe threat to biodiversity and undermines the global ambition of achieving a nature-positive future by 2050. Despite pressing calls for action, the United Nations (UN) treaty on plastic pollution failed to reach consensus at the Intergovernmental Negotiating Committee (INC)-5.1 meeting in Busan (December 2024) and again at INC-5.2 in Geneva (August 2025), where delegates deferred binding commitments. Consequently, biodegradable plastics have gained momentum as a viable alternative, with the global market projected to exceed multi-billion-dollar valuations by 2030, particularly across the Asia-Pacific, Europe, and North America. However, issues of scalability, cost, and mismatches between material performance and waste management infrastructure continue to limit their wider adoption. Although corporate initiatives that prioritize responsible sourcing and waste reduction demonstrate early progress, environmental trade-offs remain. While a global treaty is essential, immediate efforts must address the technical and economic challenges of positioning biodegradable plastics as effective and practical transitional tools. Strategic collaboration between policymakers and industry could accelerate progress toward the UN Sustainable Development Goals, aligning with the 2025 World Environment Day theme of ending plastic pollution. Bridging these gaps offers a promising pathway for mitigating ecological damage as global governance frameworks continue to evolve. Plastic pollution and the increasing economic burden of plastic waste management highlight the urgent need for circular and sustainable solutions. Although bioplastics currently represent less than 1% of global plastic production, their production is rapidly increasing, positioning them as a promising alternative to conventional plastics. Since many bioplastics can be produced from renewable resources, including agricultural residues and other bio-waste, they also offer an effective pathway for waste valorization and sustainable resource management. To further advance the bioplastics industry, greater public awareness, continued research and development, supportive policies, and strong collaboration among academia, industry, government, and other stakeholders are essential.
|
|
Scooped by
mhryu@live.com
June 22, 11:12 PM
|
Xylanases are central to lignocellulosic biomass degradation, yet current methods lack the specificity to resolve how enzymes distinguish complex xylan structures decorated with arabinofuranose (Araf) and 4-O-methyl-glucuronic acid (MeGlcA). Here, we report a suite of chemically-defined activity-based probes (ABPs) that enable the selective detection of arabinoxylan- and glucuronoxylan-specific xylanases (AXXs and GXXs). These cyclophellitol-derived ABPs covalently label retaining xylanases at their active sites, allowing precise mapping of substrate specificity across diverse glycoside hydrolase families. Crystallographic and mass spectrometric analyses reveal the molecular basis of probe selectivity, while in-gel and pull-down assays demonstrate their effectiveness in profiling xylanase activities in complex bacterial and fungal proteomes, including cellulosomes. By integrating activity-based protein profiling (ABPP) with sequence similarity networks (SSNs), we further show that xylanase specificity can be predicted from sequence alone, enabling rapid functional annotation of uncharacterized xylanases. This chemoproteomic strategy provides a powerful platform for discovering and engineering substrate-specific enzymes for biomass valorisation, microbial ecology, and biotechnological applications. Xylan is a complex plant polysaccharide with chemical decorations that shape its degradation. Here, the authors develop selective probes for xylanases targeting these structures in biological samples, offering a powerful approach for enzyme discovery and optimization for biotechnology.
|
|
Scooped by
mhryu@live.com
June 22, 10:29 PM
|
Nicotinamide adenine dinucleotide (NAD+) is one of the most important metabolic coenzymes that not only drives redox reactions and energy production but also acts as a critical substrate for several enzymes involved in immune signalling, DNA repair and epigenetic regulation. Viral infections are known as potent modulators of NAD+ metabolism, with pathogens such as SARS-CoV-2, influenza A virus, Zika virus, herpes simplex virus and human immunodeficiency virus altering NAD+ biosynthesis and consumption to benefit their persistence and replication. In this review, we summarize the current understanding of NAD+ metabolism and its regulatory enzymes: sirtuins, poly (ADP-ribose) polymerases and CD38/CD157. We then discuss the interplay between NAD+ homeostasis and virus infection. Understanding how diverse viruses manipulate NAD+ metabolism could lead to broad-spectrum antiviral strategies grounded in metabolic resilience.
|
|
Scooped by
mhryu@live.com
June 22, 10:22 PM
|
In plant synthetic biology, precise control of gene expression requires constitutive promoters with quantitatively defined transcriptional strengths. Here, we identified endogenous constitutive promoter candidates from Nicotiana benthamiana. The transcriptional activities of the selected promoters were measured using GFP-based and dual-luciferase reporter assays in plants and benchmarked against the CaMV 35S promoter, revealing substantial variation in promoter strengths. Promoter activities were further evaluated in heterologous species, including Brassica rapa, Capsicum annuum, and Zea mays, using protoplast-based dual-luciferase assays. Cross-species analyses showed that relative promoter ranking was partially retained among dicot species, whereas transcriptional activity varied substantially depending on species and was generally reduced in maize. Together, this study defines a set of endogenous constitutive promoters across a range of transcriptional strengths, providing quantitative guidance for selecting endogenous alternatives to the CaMV 35S promoter in plant biotechnology and synthetic biology applications.
|





























the fabrication of the CMA/CTA DN gels and their acid-responsive release of zhongshengmycin, Zn2+, and phenylalanine.
under acidic conditions, the protonation of the carboxylic group of Phe weakens its coordination with Zn2+, leading to the disassembly of the Phe-Zn2+ network.