Your new post is loading...

|
Scooped by
mhryu@live.com
Today, 7:01 PM
|
The rapid emergence of multidrug-resistant bacteria has created an urgent need for improved antimicrobial discovery and screening platforms. Here, we present ARCADIAMP, a generative and virtual screening platform that couples an iterative-learning discrete denoising diffusion probabilistic model with a two-stage Evolutionary Scale Modeling 2 (ESM2)-based antibacterial activity classifier to generate, classify, and prioritize potent AMPs with high activity, low toxicity, and favorable serum stability. Eight of the ten experimentally screened peptide candidates showed antimicrobial activity (MIC ≤ 32 μg/mL), while one generated candidate, Arcinin, demonstrated strong activity against ESKAPE pathogens (MIC 8–32 μg/mL), low hemolytic activity (LC50 > 512 μg/mL for human red blood cells), and strong serum-retained activity (MIC 32 μg/mL in 50% bovine serum for four ESKAPE species). Electron microscopy, membrane depolarization assays, time-kill kinetics, and molecular dynamics simulations showed that Arcinin acts through sub-microsecond insertion and penetration consistent with the behavior of other well-known AMPs. In a bacteria-infected wound murine model, Arcinin achieved a 4-log reduction in bacterial burden, which facilitated subsequent re-epithelialization and wound recovery. By framing antimicrobial discovery as an AI-assisted iterative optimization problem, ARCADIAMP links activity, toxicity, and efficacy and provides a scalable template for discovering therapeutically promising biologics. In this study, Markakis et al. use a generative AI model to design antimicrobial peptides that are both potent and safe. Their lead candidate, Arcinin, kills drug-resistant bacteria, spares human cells, and clears infection in a mouse wound model.
|
|
Scooped by
mhryu@live.com
Today, 6:42 PM
|
Transfer RNAs (tRNAs) are essential for protein synthesis and are extensively modified to ensure their structure and function. Direct RNA sequencing with Oxford Nanopore Technologies enables positional modification analysis but is challenged by tRNAs’ short length, redundancy, and dense modifications. We present QutRNA2, a scalable workflow that includes GPU-accelerated local alignment, statistical filtering, pairwise error profile comparison, and customizable visualization. Achieving up to 25-fold speed gains over CPU methods, QutRNA2 identifies enzyme-dependent modifications in nuclear- and mitochondrial-encoded tRNAs, demonstrated in high-volume human and mouse samples. This open-source solution provides a comprehensive, multiplexing-compatible framework for tRNA analysis, addressing a key gap in current tools. QutRNA2 is released under Apache-2.0 license and is available at https://github.com/dieterich-lab/QutRNA2.
|
|
Scooped by
mhryu@live.com
Today, 6:34 PM
|
Virus-induced genome editing (VIGE) has become a useful method by enabling transient delivery of gene-editing reagents; however, many viral systems face limitations in cargo size, host range, or reliance on transgenic Cas9-expressing plants. In this study, we developed a cymbidium mosaic virus (CymMV)-based VIGE platform that enables simultaneous expression of Streptococcus pyogenes Cas9 (SpCas9) and one or more guide RNAs (gRNAs) from a single viral RNA. In Nicotiana benthamiana, this system induced editing in the Phytoene desaturase (PDS) gene, with indel rates exceeding 50% within 6 days after inoculation, outperforming traditional delivery methods by about fivefold. Notably, over 80% of regenerated plants contained targeted mutations, and 82% of these were both transgene- and virus-free, including tetra-allelic knockouts directly in the M0 generation. Adding a Ruby-based visual counterselection marker enabled rapid, reliable identification of transgene-free, edited plants without antibiotic selection. When adapted to Phalaenopsis aphrodite orchids, the platform efficiently edited the PaPDS gene, achieving a 47% indel frequency at 20 days post-inoculation, with visible bleaching in leaf tissue from inoculated protocorm-like bodies. Additionally, expressing multiple gRNAs from a single CymMV replicon enabled multiplex editing in orchid tissues, demonstrating the system's versatility for complex, polyploid crops. Our findings broaden the use of VIGE in orchids and provide a reliable framework for precision plant breeding.
|
|
Scooped by
mhryu@live.com
Today, 4:59 PM
|
One-pot CRISPR diagnostics face a fundamental incompatibility: isothermal nucleic acid amplification enables rapid target accumulation, whereas CRISPR activation irreversibly consumes those substrates, destabilizing reaction kinetics. Here we show that reaction order can be programmed into DNA primers through thermodynamic design. Differences in primer-binding strength create two sequential amplification stages, delaying CRISPR activation until enough amplicons have accumulated without physical separation or external control. The design also introduces the protospacer adjacent motif (PAM), a short sequence required for CRISPR recognition, through the primer rather than relying on its presence in the native target, expanding target accessibility while retaining single-nucleotide discrimination. An ordinary differential equation model captures the threshold behavior and establishes a predictable framework for primer design. Building on this principle, we develop Thermodynamically Encoded Molecular Programming for One-pot diagnostics (TEMPO), which achieves attomolar sensitivity within 30 min and enables sequencing-concordant SNP genotyping and pathogen detection in a single-step microfluidic format. One-pot CRISPR diagnostics are limited by kinetic conflict between isothermal amplification and CRISPR detection. By thermodynamically programming reaction order into DNA primers, the authors create staged amplification enabling rapid, sensitive, single-step nucleic acid testing.
|
|
Scooped by
mhryu@live.com
Today, 4:10 PM
|
Protein thermostability is a critical property for both industrial and biomedical enzyme applications, yet experimental evaluation of mutation-induced stability changes remains laborious and costly. Here, we present ThermoFusion, a hybrid deep learning framework that integrates 3D protein structure embeddings from ThermoMPNN with sequence-based embeddings from the pretrained protein language model ESM2 to predict the effects of single-point mutations on protein stability (ΔΔG). ThermoFusion exhibits robust generalization, maintaining high predictive accuracy across out of distribution sequences with low identity to the training set -- a scenario where many other machine learning models, including ThermoMPNN and state-of-the-art tools, perform poorly due to reliance on memorization. Benchmarking on a curated enzyme dataset comprising of 105 enzymes and 3144 mutations shows that ThermoFusion reliably identifies stabilizing mutations while accurately predicting stability for enzymes beyond its training set. These results establish ThermoFusion as a powerful tool for rational enzyme design beyond its training set.
|
|
Scooped by
mhryu@live.com
Today, 4:06 PM
|
Targeted long read sequencing (LRS) of native genomic DNA (gDNA) using Oxford Nanopore Technologies (ONT) is an economically and computationally accessible method for sequencing selected genomic regions without the limitations associated with amplification-based approaches. At present, efficiency, multiplexing, and scalability remain key challenges for existing targeted LRS. We have developed Cas12a-Targeted Multiplexed Nanopore Sequencing (CTM-nSeq), which combines Cas12a-targeting, DNA fragment enrichment, and optimized adapter ligation using T7 DNA ligase. Unlike previously established protocols, CTM-nSeq is compatible with the latest ONT flow cell chemistry. Performing CTM-nSeq on a single sample with an R10.4 MinION flow cell routinely yields hundreds of on-target reads. Furthermore, CTM-nSeq enables targeting of multiple loci and is the first targeted ONT sequencing method, allowing reliable, barcode-assisted multiplexing. CTM-nSeq is an efficient and accessible method for sequencing native gDNA and analysing DNA methylation, repeat expansions, and sequence integrity. As such, CTM-nSeq has a wide range of analytical and diagnostic applications.
|
|
Scooped by
mhryu@live.com
Today, 3:31 PM
|
All organisms must allocate finite resources among growth, maintenance, and reproduction, generating trade-offs that constrain adaptation. Host-associated microbiomes are dynamic resource engines capable of generating and reallocating energy and resources for their hosts. In doing so, we argue they may recalibrate the trade-offs fundamental to host life history evolution.
|
|
Scooped by
mhryu@live.com
Today, 1:59 AM
|
Conjugative plasmids are a class of mobile genetic elements capable of efficient transfer between bacterial cells. Although they can introduce beneficial traits such as antibiotic resistance to recipients, they may also behave as genetic parasites. Bacteria would thus be expected to have evolved barriers to plasmid conjugation. However, the distribution of these barriers and their underlying mechanisms remain poorly understood. Here, we performed a large-scale analysis of 364 diverse strains of the opportunistic pathogen Acinetobacter baumannii as recipients of the broad-host-range conjugative plasmids R388 and RP4. Major variations in host susceptibilities to conjugation, with limited phylogenetic association, suggested multiple and fast-evolving plasmid-specific barriers. Functional genetic analyses revealed a role for core genes, pointing to epistasis or genetic background effects. This is illustrated by the previously unrecognized role of H-NS expression in alleviating conjugation barriers in a strain-dependent manner. Most importantly, we identified three novel immune systems protecting bacteria against conjugation by R388 and RP4. Their patchy distribution within the species, and that of their homologs across bacteria, indicate that they are part of a dynamic repertoire of immune systems against conjugation. While the Ishtar system promotes plasmid loss through putative HEPN nuclease domains, Namtar and Attar sense distinct components of the R388 type IV secretion system (T4SS) to trigger a non-proliferative, energetically depleted state, analogously to the abortive infection response of anti-phage defenses. Live imaging of conjugation showed Namtar halting cell division in E. coli recipients, conferring population-level immunity against plasmid spread via horizontal and vertical transmission. The existence of immune systems specifically targeting T4SS components suggests that conjugative plasmids impose a selective disadvantage greater than previously thought. This work reveals an additional layer of bacterial immunity directed at a class of genetic elements driving dissemination of antibiotic resistance.
|
|
Scooped by
mhryu@live.com
Today, 1:22 AM
|
The rapid rate of virus discovery renders manual curation by taxonomy experts increasingly impractical, creating a need for reliable software that can reproducibly assign viral contigs to taxa at all fifteen ranks of the virus taxonomy. We led an open community challenge for the computational taxonomic classification of viruses and assembled a dataset of virus sequences combining expert-curated and metagenomic sequences. Seventeen teams contributed a total of thirty-four automated, fully reproducible classification pipelines. Most tools correctly assigned viruses belonging to established species, genera, or families, but viruses that are unclassified at those lower ranks remain challenging. This study provides datasets, open-source software, novel approaches, and recommendations to benchmark computational taxonomic classification of viruses, and support organizing the many viruses discovered in big omics data.
|
|
Scooped by
mhryu@live.com
Today, 1:17 AM
|
Human infection often results from accidental host–pathogen interactions rather than from evolutionary microbial adaptation. In this forum, we highlight how intrinsic host plasma membrane properties—deformation, curvature sensing, receptor enrichment, and membrane reservoirs—support diverse bacterial processes of adhesion and entry.
|
|
Scooped by
mhryu@live.com
Today, 12:40 AM
|
As synthetic genomics scales toward the construction of increasingly larger genomes, computational strategies are needed to address technical feasibility. We introduce an algorithmic framework for the minimum-cost synthetic genome planning problem, aiming to identify the most cost-effective strategy to assemble a target genome from a source genome through a combination of reuse, synthesis, and join operations. By comparing dynamic programming and greedy heuristic strategies under diverse cost regimes, we demonstrate how algorithmic choices influence the cost efficiency of large-scale genome construction. In parallel, solving the minimum-cost synthetic genome planning problem can help us better understand genome architecture and evolution. Using both single closely related templates (e.g., bat coronavirus RaTG13) and diverse multisource consensus analyses, our results revealed that conserved regions such as ORF1ab can be reconstructed cost-effectively via sequence reuse. In contrast, highly variable regions such as the S (Spike) gene necessitate expensive de novo DNA synthesis. This highlights a concrete biological and economic trade-off in genome design: evolutionary sequence conservation dictates the financial feasibility of fragment reuse, whereas rapid viral adaptation incurs high synthesis penalties.
|
|
Scooped by
mhryu@live.com
Today, 12:09 AM
|
We present FLOWR.ROOT, an SE(3)-equivariant flow-matching foundation model that unifies pocket-aware 3D ligand generation with multi-endpoint binding affinity prediction (pIC50, pKi, pKd, pEC50) and pLDDT-based confidence estimation in a single backbone. One trained model supports de novo pocket-conditional generation, interaction- and pharmacophore-conditional sampling, scaffold hopping and elaboration, and fragment growing or replacement, enabled by a mixed isotropic–anisotropic prior placement strategy. Training proceeds in three stages: large-scale pre-training on billions of ligand conformations and millions of mixed-fidelity protein–ligand complexes, refinement on curated co-crystal data, and project-specific adaptation via parameter-efficient LoRA finetuning. Joint structure–affinity modelling enables inference-time importance-sampling guidance for single- and multi-objective design without external scoring functions. Case studies on kinase selectivity (CK2α/CLK3) and scaffold elaboration on TYK2, ERα, and BACE1 illustrate utility from hit identification through lead optimization. Structure-based generative modeling is rapidly reshaping drug discovery by enabling pocket-aware ligand design alongside predictive evaluation of binding properties. This manuscript introduces FLOWR.root, an SE(3)-equivariant flow-matching framework that jointly generates high-quality 3D ligands and predicts multi-endpoint binding affinities, demonstrating state-of-the-art performance, efficient domain adaptation, and practical impact across de novo design, scaffold elaboration, and lead optimization workflows.
|
|
Scooped by
mhryu@live.com
July 6, 2:42 PM
|
Autonomous self-reproduction is a major goal of bottom-up synthetic biology aimed at building artificial cells. This requires that the genome be replicated by its self-encoded replication machinery. While the reconstituted E. coli chromosomal replication system, termed the Replication-Cycle Reaction (RCR) system, offers a promising platform for genome-scale replication, its generation from genetic information has not yet been achieved. Here we show that a 53 kb circular DNA, termed RCR module-genome, encoding all 26 RCR proteins, can self-replicate in a one-pot reaction when expressed using the protein synthesis using recombinant elements (PURE) system. We first built a prototype of the RCR module-genome and then optimized reaction conditions and solved expression bottlenecks to achieve robust self-replication. This artificial module-genome supports more than 28 doublings of recursive self-replication. This system, termed PRIMES (PURE-driven RCR for In-vitro Module-gEnome Self-replication), represents a milestone toward constructing self-reproducing artificial cells.
|
|
|
Scooped by
mhryu@live.com
Today, 6:57 PM
|
Bacteria residing in biofilms are embedded in an extracellular matrix. Whereas biofilm formation is well studied, less is known about biofilm dispersion, although enzymatic extracellular matrix degradation is suspected to play a key role. Here we show that Bacillus subtilis biofilms can alternatively eject a specific cell type, locally and anisotropically, using mechanical forces arising from a self-generated hydrogel. Single-cell resolution imaging combined with mathematical modelling, and chemical and genetic perturbations, show that the production of the extracellular poly-γ-glutamic acid (γ-PGA) polymer is necessary to drive this cell ejection. Specifically, osmotic pressure from the γ-PGA hydrogel propels interior cells through the outer layers to break free from the biofilm. We demonstrate control over this process through γ-PGA modulation such that biofilm dispersion can be either inhibited or promoted. Forceful ejection driven by γ-PGA has so far only been described in marine organisms such as jellyfish. Our discovery of biofilm cell ejection via γ-PGA thus reveals not only a previously uncharacterized biofilm dispersion mechanism but also an unexpected mechanistic parallel to evolutionarily distant Cnidaria. Bacillus subtilis biofilms locally disperse by ejecting motile cells through osmotic pressure generated by their poly-γ-glutamic acid hydrogel, a mechanism resembling that through which Cnidaria eject nematocysts.
|
|
Scooped by
mhryu@live.com
Today, 6:38 PM
|
D-amino acids (DAAs), once considered minor enantiomers, are now recognised as abundant and dynamic components of marine and terrestrial organic matter. While they do not participate in ribosomal protein synthesis, they play crucial roles in microbial physiology, particularly in bacterial cell wall structure. This review systematically synthesises the current understanding of DAA sources, distribution and fate, with a central focus on the microbial catabolic pathways that drive their recycling in marine and terrestrial environments. We show that, despite differences in DAA distribution and bioavailability between marine and terrestrial ecosystems, the core catabolic strategies adopted by bacteria—conversion to α-keto acids, L-amino acids (LAAs) or Gly—are largely conserved. We also evaluate limitations in current studies and major knowledge gaps, including the unclear role of marine fungi in DAA turnover and the relative lack of systematic studies on DAA-catabolising taxa and pathways in terrestrial microbial communities. This review highlights the ecological significance of microbial-mediated DAA recycling in marine and terrestrial environments, offering a better understanding of the global biogeochemical cycling of DAAs.
|
|
Scooped by
mhryu@live.com
Today, 6:22 PM
|
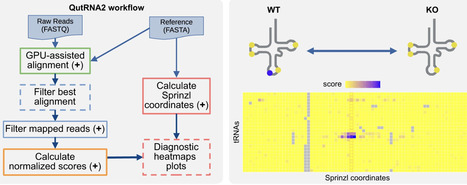
The functional diversification of O2-tolerant [NiFe]-hydrogenases using orthogonal translation systems (OTSs) offers a promising strategy for developing advanced biocatalysts and biohybrid energy platforms. However, plasmid-based OTSs frequently impose metabolic burdens and suffer from plasmid instability during fermentation, particularly when co-produced with complex metalloenzymes. To overcome these bioprocess limitations, we employed CRISPR/Cas9-mediated genome editing to integrate a psychrophilic pyrrolysyl-tRNA synthetase/tRNA pair into the E. coli BL21 genome. The resulting strain provided a plasmid-free orthogonal translation background that supported amber suppression-mediated expression of the regulatory [NiFe]-hydrogenase (RH) of Cupriavidus necator. Using this genomically integrated OTS, we achieved the production of a full-length, catalytically active RH variant. Our results demonstrate that chromosomal OTS is compatible with the efficient production and maturation of complex metalloenzymes. The present work lays the groundwork for the bio-orthogonal engineering of hydrogenases and related hybrid biocatalysts.
|
|
Scooped by
mhryu@live.com
Today, 4:45 PM
|
Rapid and reliable quantification of bacterial dynamics at the cellular level is critical for pathogen sensing, live-dead bacterial assays, and monitoring of bacteria fitness and viability. Here, we demonstrate bacterial fitness quantification by capturing individual cells on topological defects of micro-scale liquid crystal emulsion droplets. The emulsion droplets are composed of phase-separated nematic liquid crystal and fluorocarbon components and exhibit an asymmetric mass distribution. A topological singularity in the director field of the liquid crystal phase localizes tailormade surfactants that tether a single bacterium per droplet. Active motion of the bacterium induces a tilt and azimuthal rotation of the droplet trap, which is counteracted by gravity acting on the droplet center of mass. By comparing the observed dynamics of a tethered bacterium’s stochastic movement to a computational model of bacterial motion on spherical surfaces that is based on the classical Ornstein-Uhlenbeck process, we quantify the fitness of bacteria subjected to starvation over several days. This pathogen fitness sensing concept, which relies on the scalable chemical design of single bacterial cell traps, a robust optical readout, and a theoretical understanding of bacterial dynamics on spherical surfaces, offers opportunities for rapid pathogen activity assessment, micro-biological sensing, and biologically powered micro-actuator systems. The ability to rapidly and accurately assess bacterial fitness is crucial for effective pathogen detection and monitoring. Here, authors develop a method using microscale liquid crystal emulsion droplets to capture individual bacteria, enabling real time quantification of their fitness through the analysis of droplet dynamics influenced by bacterial motion.
|
|
Scooped by
mhryu@live.com
Today, 4:08 PM
|
Transcriptional termination efficiency is considered an important parameter for fine tuning bacterial gene expression. Still, the design principles that determine transcription termination efficiency remain poorly understood. In this study, we aimed to investigate the impact of the 3′ untranslated region (3′UTR) on gene expression in Escherichia coli and other bacteria. First, 3′UTR variant sequences were generated, with randomized 30 bp sequences inserted between the STOP-codon and an intrinsic terminator, consisting of a GC-rich hairpin and a downstream poly(U)-tail. Using three reporter genes, it was found that different 3′UTR sequences resulted in an up to five-fold difference in protein production, independent of the upstream coding sequence. The highest protein production was achieved when an adenosine was present directly upstream of the terminator hairpin. This was consolidated by systematic substitution of key nucleotides of the terminator and assessing their effect on mRNA and protein levels. Subsequently, we developed a predictive random forest machine learning model trained on the termination efficiency of different natural and synthetic terminator sequences, revealing an important role for the nucleotides directly upstream of the terminator hairpin. Altogether, this study showed that an additional adenosine nucleotide upstream of the terminator hairpin leads to improved protein production while reducing terminator read-through.
|
|
Scooped by
mhryu@live.com
Today, 4:00 PM
|
Here we introduce OpenEvo, a fully open-source, low-cost turbidostat platform for automated continuous culture and directed evolution experiments. Existing tools are expensive, complex, or lack open-source hardware; OpenEvo addresses this gap. OpenEvo is a complete, fully automated evolution platform with detailed, illustrated construction instructions for beginners, open-source software and firmware, and a single device priced around $300. An optional PC-based version offers enhanced functionality, including remote access, programmable evolution cycles, programmable LED stimulation, and a data visualization tool. OpenEvo can cycle through three types of media for positive, negative, and neutral selection conditions, supporting a wide range of experimental designs. We validate the use of OpenEvo by evolving H. volcanii to grow from 15% to 12% salt over ~150 cycles, ~1,000 hours. Evolved cells grew 36% faster than wild-type at 12% salt. Whole-genome sequencing of adapted cells found SNPs and large deletions. We also demonstrate positive and negative selection using the OpenEvo LEDs to drive optogenetics via a Phytochrome B-based optogenetic tool, with light as the selection stimulus during over 4000 hours of growth. OpenEvo lowers the technical and cost barriers for continuous evolution experiments, serves as a teaching tool, and is designed to grow an open community of users who share modifications.
|
|
Scooped by
mhryu@live.com
Today, 3:11 PM
|
Lead (Pb) and cadmium (Cd) remain among the most persistent and hazardous heavy-metal contaminants in industrial effluents, posing severe risks to ecosystems and human health due to their non-biodegradable nature and high toxicity. In response to the limitations of conventional chemical remediation technologies, this study evaluates the potential of E. coli K-12 MG1655 to function as a microbially driven system for the detoxification, sequestration and recovery of Pb and Cd. Emphasis is placed on oxalic acid production as a mechanistic basis for metal tolerance. HPLC confirmed that E. coli K-12 MG1655 synthesizes oxalic acid under metal stress, with Pb exposure eliciting the better oxalate output, providing evidence consistent with metal–oxalate-associated detoxification during metal stress. Bioaccumulation studies using inductively coupled plasma–optical emission spectrometry revealed exceptional metal removal efficiencies, reaching 99.94% for Pb and 97.77% for Cd at 1,000 p.p.m., while Pb+Cd mixed-metal systems maintained high overall uptake (98.19%). These results demonstrate that E. coli can sequester metals across a wide concentration range with minimal inhibition from competitive ion interactions. Metal recovery from loaded biomass was evaluated through acid desorption and ohmic heating. Nitric acid (0.1 M HNO3) achieved the highest recovery efficiencies (Pb, 98.5%; Cd, 91.5%), whereas ohmic heating yielded moderate (Pb, 45.38%; Cd, 45.83%) but environmentally favourable recovery without chemical additives. The integrated findings illustrate a complete microbial bioremediation–recovery cycle encompassing detoxification via oxalic acid, high-efficiency metal sequestration and effective downstream recovery. This integrative study establishes E. coli K-12 MG1655 as a promising candidate for closed-loop bioremediation systems linking detoxification, sequestration and recovery of heavy metals.
|
|
Scooped by
mhryu@live.com
Today, 1:51 AM
|
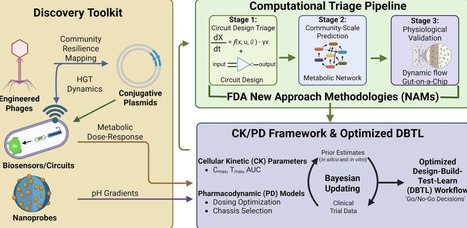
Despite the vast opportunities for therapeutic manipulation of the gut microbiome, recent late-stage clinical failures of engineered live biotherapeutic products (eLBPs) highlight critical knowledge gaps in ecological barriers and community dynamics. In this review, we propose repurposing the current eLBP toolkit as a set of discovery instruments that yield quantitative outputs for predictive modeling. We examine cutting-edge approaches in microbiome engineering and outline opportunities for their use in tandem with systems engineering methodology to conduct functional probing that establishes quantitative parameters describing community resilience, metabolic flux, and host-microbe interactions. Next, in light of FDA guidance on New Approach Methodologies, we detail how in silico and in vitro modeling approaches can be combined and leveraged not only for a priori triage of unviable designs, but can also be integrated into design-build-test-learn (DBTL) pipelines for functional forecasting. Building off an emerging cellular kinetics/pharmacodynamics (CK/PD) framework, we develop a Bayesian updating workflow that encapsulates eLBP-adapted equivalents of pharmacological parameters such as Cmax, Tmax, and AUC. Further, we adapt this framework for adaptive or prospective use, rather than purely retrospective application, supporting trial design rather than post-hoc analysis. This approach repositions eLBP development from an empirical, intuition-based process toward a predictive, model-informed pipeline that aligns with emerging regulatory frameworks.
|
|
Scooped by
mhryu@live.com
Today, 1:20 AM
|
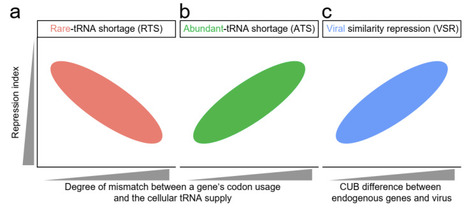
Viral infection induces tRNA competition between viral and host genes, often repressing host translation. However, how endogenous genes are affected by this competition remains unclear. Three possible hypotheses are considered: abundant-tRNA shortage, rare-tRNA shortage, and viral similarity repression. Pan-virus Ribo-seq data show that endogenous genes with codon usage bias (CUB) matching host tRNA supply or viral CUB are strongly repressed, due to a positive correlation between endogenous CUB-tRNA mismatch and endogenous-viral CUB difference, supporting the abundant-tRNA shortage and viral similarity repression hypotheses. In E. coli experiments with synonymous gentamicin resistance proteins, this positive correlation supports abundant-tRNA shortage, while a non-positive correlation supports rare-tRNA shortage, and both positive and non-positive correlation types support viral similarity repression. Finally, analysis of human virus genomes reveals this positive correlation for most viruses, but a non-positive correlation in a few, reflecting diverse virus–host interaction strategies. These findings establish viral similarity repression as a universal principle, uncovering previously unrecognized complexity in virus–host coevolution.
|
|
Scooped by
mhryu@live.com
Today, 12:41 AM
|
web tool, List of the web servers included in the 2026 NAR Web Server Issue
|
|
Scooped by
mhryu@live.com
Today, 12:24 AM
|
Selecting between axenic strains and microbial consortia remains a key challenge in microbial bioremediation. Here, we established a systematic quantitative framework to compare their degradation performance and biological traits across six pollutant classes: petroleum hydrocarbons, antibiotics, pesticides, polycyclic aromatic hydrocarbons (PAHs), heavy metals, and plastics, under comparable experimental contexts. For complex substrates requiring sequential transformation and mineralization, especially petroleum hydrocarbons and PAHs, microbial consortia generally showed higher degradation efficiency through metabolic division of labor, biofilm enrichment, and functional redundancy. Reported efficiency improvements ranged from approximately 17% to 73%, with selected cases reaching up to 200%. In contrast, for certain antibiotics and pesticides, consortium performance was inconsistent and sometimes inferior to acclimated or engineered axenic strains due to interspecific antagonism, metabolic inhibition, or unstable functional coordination. Based on the assembled evidence, we propose a decision-making framework for remediation strategy selection: microbial consortia are preferable for complex, multi-step substrates, whereas axenic strains are more suitable for highly toxic contaminants or scenarios requiring strong operational controllability. This study provides quantitative evidence and practical guidance for rational microbial strategy selection, system design, and scale-up in bioremediation engineering.
|
|
Scooped by
mhryu@live.com
July 6, 5:02 PM
|
Mutagenesis is a fundamental, yet poorly understood, source of genetic variation that underpins microbial evolution and adaptation. When the Bacillus subtilis replicative DNA polymerase (DNAP) PolC encounters DNA lesions induced by endogenous or exogenous insults, it stalls and disassembles. If error-free DNA damage tolerance (DDT) sub-pathways fail to circumvent the lesion, bipartite translesion synthesis (TLS) DNAPs (PolY1 or PolY2 together with PolA) may bypass the damage to resume DNA synthesis. Mismatch repair subsequently removes misincorporated nucleotides. Here, we investigate which proteins loaded at stalled replication forks influence mutation dynamics mediated by TLS DNAPs. We demonstrate that ΔrecA, ΔpolA, or ΔpolY1 ΔpolY2 mutations strongly reduce cell survival following DNA damage and mutagenesis. The accessory proteins DisA, RarA, RecD2, DinG, and Mfd, which physically interact with PolA and/or RecA, differentially affect cell survival after DNA damage in the absence of TLS DNAPs and differentially modulate mutagenesis by regulating the activity of distinct TLS DNAPs, highlighting their roles in error-prone DDT. We also reveal that SOS-independent mutagenesis operates in the ΔpolA ΔrecA background. Elucidating the regulatory network underlying TLS provides a framework to understand bacterial speciation and may uncover new avenues to limit antibiotic resistance emergence.
|



















![Genomically integrated orthogonal translation system in Escherichia coli enables production of functional modified [NiFe]-hydrogenases | Mcf | RMH | Scoop.it](https://img.scoop.it/5Ngfoy5n83nHWAnvyTszJTl72eJkfbmt4t8yenImKBVvK0kTmF0xjctABnaLJIm9)