 Your new post is loading...

|
Scooped by
mhryu@live.com
Today, 1:49 AM
|
Arbuscular mycorrhizal fungi form symbioses with ~70% of plant species, building hyphal networks that exchange nutrients for host-derived carbon. These tubular networks move ~1 billion metric tons of carbon per year into Earth’s soils. However, we have no quantitative understanding of the hyphal infrastructure required to carry out this resource transfer. We assembled data from 322 studies representing more than 16,000 soil cores across nine biomes and developed machine-learning models to predict hyphal densities globally. With robotic imaging of more than 300,000 hyphae, we calibrated a biomass model from our spatial predictions. We estimate that global topsoils contain 1.10 × 1017 ± 0.13 × 1017 SD kilometers of living hyphae, weighing ~300 ± 60 SD megatons, ~4- to 6-fold the biomass of humans. Our uncertainty analyses identified undersampled ecosystems that require additional empirical attention.
|
|
Scooped by
mhryu@live.com
Today, 1:37 AM
|
Messenger RNA (mRNA)-based therapeutics have emerged as a new class of biological medicines, clearly exemplified by the global deployment of mRNA vaccines against the COVID-19 pandemic. Currently, therapeutic mRNA is primarily produced through in vitro transcription that suffers high production costs. Until now, intracellular manufacture of mRNA has been challenging due to the presence of ubiquitous RNases in vivo. Here, we have developed a new approach that protects eukaryotic mRNA from RNase degradation ensuring longevity and integrity of mRNA inside microbial cells. Through targeted strain and molecular engineering, our approach involves specially designed inserts in mRNA that facilitate formation of stabilized and protected protein-mRNA complexes. In addition to vastly improved stability, the protein-mRNA complexes enable convenient purification of mRNA from cell lysate with high purity using conventional chromatography. The work reported here promises a scalable, rapid, and low-cost approach to produce fully functional eukaryotic mRNA using well-known microbial systems.
|
|
Scooped by
mhryu@live.com
Today, 1:20 AM
|
By leveraging the capacity of near-infrared light to penetrate deep tissue, we devised an engineered bacterial system for fluorescence imaging-guided photothermal/gene combined cancer therapy. By incorporating a thermosensitive plasmid carrying a payload of cytolysin A (ClyA) into the probiotic E. coli Nissle 1917 (EcN) and incubating it with the photothermal agent indocyanine green (ICG)/Cremophor EL, we generated engineered bacteria capable of photothermal conversion. Upon irradiation with a near-infrared laser, ICG within the engineered bacterial cells converted them into localized heat above 42°C. This simultaneous photothermal therapy cleverly activated the expression of the therapeutic protein ClyA, resulting in the synergistic killing of tumor cells. Importantly, as a natural immune adjuvant, EcN enhanced the body’s immune response. In addition, the engineered bacteria exhibited significant cytotoxic effects on tumor cells and suppressed tumor growth. Our study demonstrates a near-infrared laser-controlled bacterial platform that can be applied to precision cancer therapeutics.
|
|
Scooped by
mhryu@live.com
Today, 12:39 AM
|
CRISPR-Cas systems often rely on collateral cleavage of nucleic-acid substrates to combat recognized mobile genetic elements. Of the CRISPR-associated (Cas) RNA-guided effector nucleases, Cas12a2 stands out as the only known example exhibiting rapid collateral cleavage of three distinct substrates: single-stranded (ss)RNA, ssDNA, and double-stranded (ds)DNA, after activating upon binding cognate RNA. However, little is known about the underlying mechanisms of collateral cleavage. Here, we show, using enzyme kinetics and inhibition assays, that Cas12a2 preferentially cleaves collateral DNA over RNA substrates, even when RNA substrates are more abundant. Additionally, using enzyme mutants, enzyme kinetics, and plasmid cleavage assays, we determine that the dsDNA cleavage mechanism relies on the ‘aromatic clamp’ residues that stabilize unwound and distorted dsDNA in the RuvC nuclease active site. Leveraging the cleavage preference for collateral DNA, we demonstrate that RNA-activated Cas12a2 can readily cleave a ssDNA probe in the presence of high concentrations of non-target RNA, while an RNA-targeting Cas13a cannot. This work provides foundational kinetic and biochemical insights into the collateral cleavage mechanism and substrate preferences of Cas12a2, with immediate implications for understanding Cas12a2-based immunity and developing Cas12a2-based technologies.
|
|
Scooped by
mhryu@live.com
June 16, 11:45 PM
|
Bacterial extracellular polysaccharides play a crucial role in mediating pathogen-host interactions and bacterial fitness via biofilm formation. The Wzx/Wzy-dependent pathway is the most prevalent and conserved strategy for polysaccharide biosynthesis. Psl (polysaccharide synthesis locus) is a key biofilm matrix polysaccharide in Pseudomonas aeruginosa PAO1, and its biosynthesis machinery is predicted to be a Wzx/Wzy-dependent biosynthesis system. Here, using Psl in PAO1 as a model strain, we determine the cryo-EM structures of the PslD-PslE complex. These structures reveal that PslD-PslE complex forms a continuous, protected conduit across the entire cell envelope. Further structural and functional analyses demonstrate that the polymerase PslJ, is likely localized in the membrane lumen formed by the octameric arrangement of PslE’s transmembrane helical pairs. We propose a mechanistic model in which Und-PP-linked pentasaccharide units of Psl access PslJ through side portals in the PslE octamer, shielding the polymerization and translocation processes from degradation by PlsG, a periplasm-localized endoglycosidase. The iterative addition of incoming repeat units to the reducing terminus of the growing polysaccharide chain is hypothesized to drive Psl export through the channel, a mechanism that may be conserved across the Wzx/Wzy-dependent polysaccharide biosynthesis pathways. The mechanism of Psl polysaccharide assembly and export to the cell surface is still unclear. Here the authors determine the cryo-EM structures of PslD-PslE complex, providing insights into Psl polysaccharide biosynthesis.
|
|
Scooped by
mhryu@live.com
June 16, 10:59 PM
|
Natural products such as polyketides and nonribosomal peptides (NRPs) are important sources of bioactive compounds, including many antibiotics. Many of them are assembled by modular enzyme complexes and further modified and diversified by tailoring reactions encoded by biosynthetic gene clusters (BGCs). Although natural products and their coding BGCs describe different data modalities of the same biochemical process, a unified language to jointly describe their biochemistry is lacking. Here we introduce a sequence-based representation of the core biosynthesis of modular natural products, which we call primary sequences, that bridges chemical structures and BGCs. We also present RetroMol, an algorithm that parses either natural product structures or their encoding BGCs into their primary sequences of natural product building blocks. RetroMol allows for similarity scoring between natural products and BGCs, enabling the retrieval of compounds, BGCs, and a combination of the two, based on their biosynthetic similarity. This can, for instance, be used to retrieve biosynthetically similar but structurally dissimilar compounds, or link natural products to candidate coding BGCs in large experimental datasets. We demonstrate the latter by rediscovering the nocardichelin B BGC as a proof of principle. We also exemplify the utility of biosynthetic similarity by showing various pairs of biosynthetically similar compounds with low structural similarity. Together, these results establish primary sequences as a shared biosynthetic encoding for natural product comparison and BGC prioritization.
|
|
Scooped by
mhryu@live.com
June 16, 10:19 PM
|
Primer bias in 16S rRNA gene amplicon sequencing can distort microbial diversity estimates by underrepresenting key taxa. We introduce a modified primer pair (V4-EXT) targeting the hypervariable V4 region of bacterial and archaeal 16S rRNA genes, with improved in silico taxonomic inclusivity. To benchmark performance, we analyzed 938 samples from terrestrial, aquatic, and host-associated habitats, comparing microbial community profiles derived with V4-EXT and the currently most widely used V4-targeted primers. V4-EXT substantially improved the detection of Patescibacteria and other underrepresented lineages, such as Chloroflexi and Iainarchaeota, while enhancing recovery of novel amplicon sequence variants across sample types. Overall, V4-EXT provides broader taxonomic coverage and more inclusive microbial community profiles, particularly in high-diversity ecosystems such as groundwater and soils. We propose V4-EXT as a robust successor for comprehensive microbial community analysis across diverse habitats.
|
|
Scooped by
mhryu@live.com
June 16, 9:54 PM
|
Statistical and culture-based models propose that environmental disturbances reshape competitive interactions among functionally redundant microbial taxa. However, the mechanisms driving these changes and their impact on biogeochemical processes remain largely untested in soil, owing to the challenge of linking functions to specific taxa in highly diverse and functionally complex soil microbiomes. Here, we simulated environmental disturbance in microcosms containing organic carbon-rich or sandy soils. Using bacterial and archaeal nitrifiers, a functionally tractable microbial guild, we examined how disturbance restructures competition among diverse microbial taxa within this guild. In both soils ammonia-oxidizing archaea (AOA) predominated numerically and functionally under steady climax conditions. Following disturbance, ammonia-oxidizing bacteria (AOB) and complete ammonia oxidizers (comammox) rapidly gained a growth-related competitive advantage, likely due to increased per-cell ammonium availability supporting their intrinsic high growth rates. AOB recolonization was essential for full post-disturbance nitrogen turnover, resulting in elevated nitrification rates and increased nitrous oxide emissions. Nitrification rate did not fully recover when AOB were inhibited. In contrast, AOA and comammox played a dispensable role in recovering post-disturbance nitrification, limited by slower growth and lower per-cell activity, respectively. Competitive regrowth ability of microbial species showed a tradeoff with pre-disturbance abundance, highlighting the enhanced post-disturbance role of rare-abundance AOA, in addition to AOB phylotypes. Our findings demonstrate that bacterial and archaeal nitrifiers constitute a continuous spectrum between r- and K-strategists. Disturbance reshapes competition through differential growth and activity traits among functionally redundant taxa, favoring AOB and thereby transforming community assembly while intensifying nutrient cycling and greenhouse gas fluxes.
|
|
Scooped by
mhryu@live.com
June 16, 5:56 PM
|
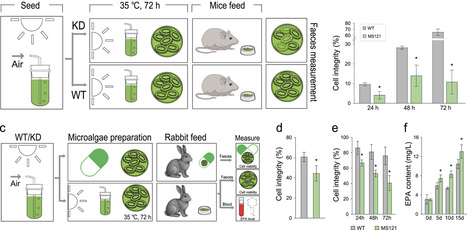
Microalgae are promising photosynthetic platforms for high-value compounds, yet their industrial use is often hindered by a trade-off between robust growth and the metabolic burden of payload production or cell wall disruption. Constitutive engineering for these traits compromises cultivator fitness. Here we report the development of a versatile, thermal-regulated “PLUG-IN” chassis in Nannochloropsis oceanica that enables programmable control of metabolic output and cell integrity. Comparative transcriptomics identify two highly heat-inducible promoters (PNoED and PNoUK), which we use to construct a modular thermal gene-amplification. Heat-activated AtWRI1 expression enhances triacylglycerol and eicosapentaenoic acid accumulation, while temperature-dependent silencing of the cellulose synthase gene CesA1 triggers rapid cell-wall weakening without affecting growth under permissive conditions. Coupling metabolic and cell-wall modules yields strains capable of grind-free lipid recovery and substantially improved intracellular product accessibility. Notably, these engineered thermally controlled programs remain functional in mammalian hosts, demonstrating cross-kingdom compatibility. This work establishes a “plug-in” chassis compatible with mammalian systems that synchronizes growth, production, and cell-wall re-configuration, providing a versatile platform for photosynthetic bioproduction and microalgal synthetic biology.
|
|
Scooped by
mhryu@live.com
June 16, 12:57 AM
|
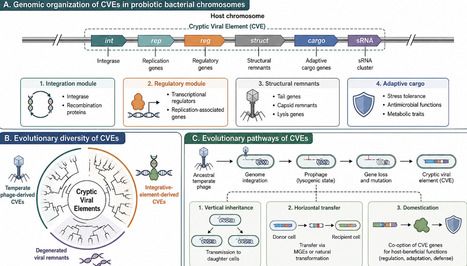
Cryptic viral elements (CVEs) are virus-derived genomic sequences integrated into bacterial chromosomes that have lost the ability to form infectious particles but are retained through evolutionary processes. Emerging evidence indicates that some CVEs are functionally active rather than inert, contributing to stress adaptation, gene regulation, and genome stability in certain bacterial systems, including probiotic genera such as Lactobacillus and Bifidobacterium. For instance, defective prophage regions have been associated with oxidative stress tolerance and persistence under nutrient-limited conditions, although these roles remain context- and strain-dependent. This review critically examines the origin, diversity, and functional relevance of CVEs in probiotic genomes, distinguishing experimentally supported functions from inferred roles in metabolic flexibility and horizontal gene transfer. Current bioinformatic approaches for CVE detection are evaluated, with emphasis on their limitations, including sequence degeneration and annotation biases. While CVEs have been proposed as potential tools for strain improvement and microbiome modulation, direct experimental validation remains limited. Key challenges include incomplete functional characterization and unresolved biosafety concerns. Addressing these gaps will be essential to assess the feasibility of leveraging CVEs in applied microbiology and biotechnology.
|
|
Scooped by
mhryu@live.com
June 16, 12:30 AM
|
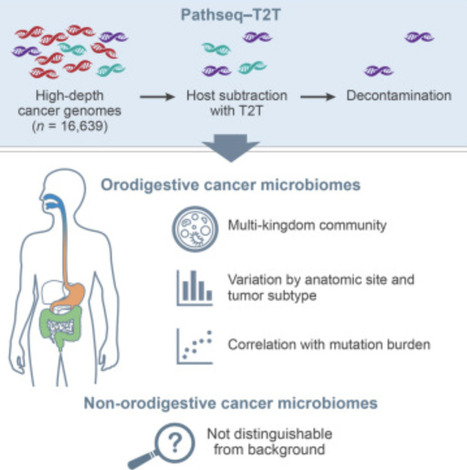
Microorganisms represent an important component of the tumor microenvironment, but conflicting reports have left the extent of microbial prevalence across cancer types unclear, necessitating more robust methods for characterizing tumor-associated microbiomes. We built and benchmarked a host-subtraction and classification pipeline to identify microbiota in whole-genome sequencing data and applied it to 16,369 high-depth tumor whole genomes from the UK 100,000 Genomes Project. After decontamination, microbial signatures were indistinguishable from the background in most cancer types. However, in orodigestive tumors, we detected multi-kingdom polymicrobial communities, including bacteria, fungi, viruses, archaea, and, in some cases, Trichomonas, a protozoan parasite. These communities varied by tumor site and subtype, with increased microbial colonization of microsatellite-instable and polymerase ε (POLE)/polymerase δ (POLD1)-mutated tumors, supported by a correlation between microbial load and tumor mutation burden observed across orodigestive cancers. This analysis helps to resolve pan-cancer microbial structure and links the tumor microbiome to host phenotype and tumor genomic context.
|
|
Scooped by
mhryu@live.com
June 16, 12:22 AM
|
Bacterial pathogens commonly become drug resistant via horizontal acquisition of antimicrobial resistance genes (ARGs), which are often encoded on mobile genetic elements (MGEs). Although bacterial defence systems are typically considered barriers to horizontal gene transfer (HGT), previous studies revealed that bacteria with more restriction-modification (RM) systems (the most abundant bacterial defences) frequently carry more MGEs. It was suggested that this counterintuitive relationship might result from stronger selection for RM systems when exposure to costly MGEs increases. Here, we test this hypothesis using a combination of modeling and bioinformatics analysis of >40,000 bacterial genomes to better understand how eco-evolutionary feedbacks between selection for RM and acquisition of MGEs shape bacterial genome evolution. Our model predicts negative associations between HGT and RM, but only if RM diversity is high. By contrast, at low RM diversity, eco-evolutionary feedbacks drive the emergence of positive associations between HGT and RM. Consistent with these predictions, we identified negative relationships between acquired ARG counts and RM counts across species but positive relationships within individual species. Collectively, our work helps to understand how RM systems shape patterns of HGT of ARGs, which may offer opportunities for targeted surveillance of strains at higher risk of horizontally acquiring novel drug resistance alleles.
|
|
Scooped by
mhryu@live.com
June 15, 11:41 PM
|
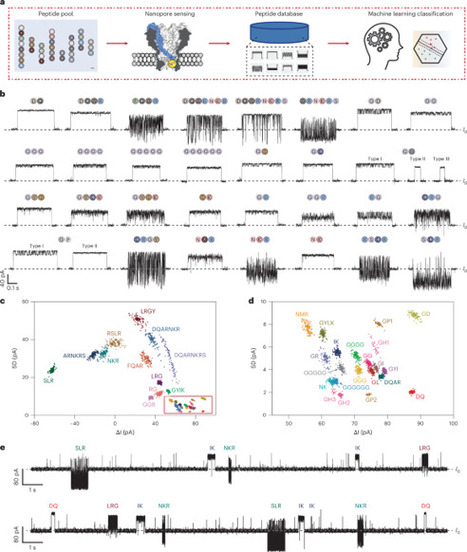
Nanopores have been explored as a potential platform for protein analysis following the success of nanopore nucleic acid sequencing. However, protein sequencing remains technically challenging and has not yet been established for proteomics use. Here a nickel-immobilized Mycobacterium smegmatis porin A (MspA-NTA-Ni) nanopore is shown to enable the identification of a range of proteomic analytes, including amino acids and peptides up to 39 amino acids in length. Under identical conditions, signals corresponding to 20 proteinogenic amino acids, 4 post-translationally modified amino acids, 32 peptides, 6 modified peptides, 11 bioactive peptides and 2 neoantigen peptides were recorded. Machine-learning-based analysis enabled classification of these analytes with a validation accuracy of up to 97.4% within the studied dataset. The MspA-NTA-Ni nanopore supports both direct peptide identification and peptide profiling following enzymatic hydrolysis. As a proof of concept, a reference peptide was digested using exo- and endopeptidases to generate overlapping peptide fragments. Nanopore measurements combined with machine learning predictions enabled the identification of fragment compositions and partial sequences, allowing reconstruction of the original peptide sequence. This hydrolysis-based approach shows sensitivity to sequence alterations, including mutations, deletions and post-translational modifications, indicating potential utility for targeted peptide characterization. A nickel-modified nanopore enables simultaneous, unambiguous identification of amino acids and peptides. Direct analysis or hydrolysis-based sequencing can be used for profiling and reconstruction, and detect single-residue mutations and post-translational modifications.
|
|
|
Scooped by
mhryu@live.com
Today, 1:44 AM
|
The human body hosts a vast and dynamic microbial ecosystem that plays essential roles in immunity, metabolism, and tissue function. Growing scientific and consumer interest in the human microbiome has accelerated innovation in topical skincare, particularly in the emerging category of probiotic, prebiotic, and postbiotic formulations. However, despite rapid market growth, most commercial products rely on non-viable microbial derivatives rather than live, strain-identified organisms, largely due to formulation, regulatory, and stability challenges. This review provides a microbial formulation perspective on probiotic skincare, beginning with an overview of the human and skin microbiomes, followed by a discussion of how beneficial microorganisms influence skin appearance and barrier function. We clarify the definitions and scientific distinctions among probiotics, prebiotics, and postbiotics and evaluate current cosmetic applications. We then explore the technical and regulatory hurdles associated with incorporating live microbes into cosmetic products, including preservative compatibility, viability, packaging, and activation requirements. Finally, we present some available strategies and technologies in an effort to keep probiotic viable until use and outline future research considerations needed to advance authentic, evidence-based probiotic skincare.
|
|
Scooped by
mhryu@live.com
Today, 1:27 AM
|
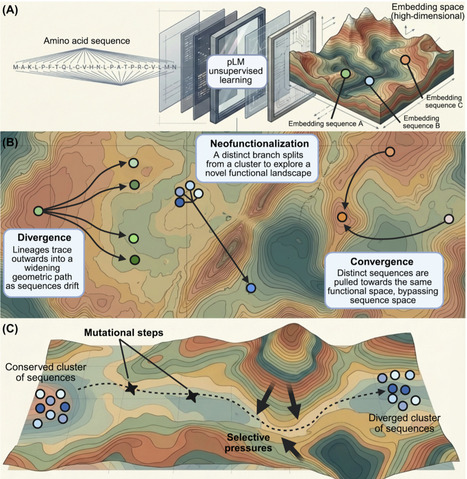
Protein language models compress protein sequences into high-dimensional embeddings that capture biochemical, structural, and functional constraints without explicit supervision. We highlight that these embeddings encode rich evolutionary information, enabling new geometry-based views of homology, divergence, and convergence, and calling for a synthesis between classical molecular evolution and systematic evolutionary embedding analysis.
|
|
Scooped by
mhryu@live.com
Today, 1:11 AM
|
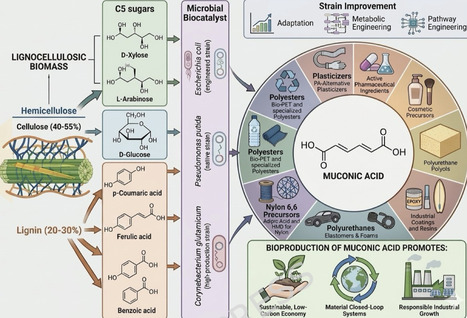
Muconic acid (MA) is characterized by two reactive carboxylic acid groups and two conjugated double bonds, making it a highly valuable industrial platform chemical with significant market potential. It serves as a key intermediate in the manufacturing of important commercial chemical products such as adipic acid and terephthalic acid. The finite fossil-based resources and climate issues due to CO2 emission have necessitated the microbial routes for the production of MA, a potential alternative to fossil-based synthesis. This review provides a comprehensive overview of recent progress in the biological production of MA. The article begins with an outline of the present catalytic routes and known biochemical pathways for MA biosynthesis. The review then focuses on metabolic engineering strategies employed in various microbial hosts including E. coli, Corynebacterium glutamicum, and Pseudomonas putida to enhance MA production from diverse feedstocks such as sugars, aromatic compounds, and lignin-derived substrates. Special attention is given to pathway optimization, host tolerance, and strategies enabling efficient conversion of lignin-derived intermediates within integrated biorefinery frameworks. Key challenges associated with scaling up bio-based MA production to industrial levels are discussed, along with potential strategies for developing robust and efficient microbial cell factories. The review concludes with future perspectives and recommendations to accelerate research progress and development in this field.
|
|
Scooped by
mhryu@live.com
June 16, 11:50 PM
|
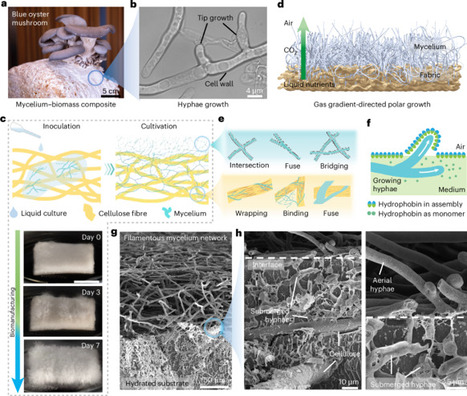
Water scarcity and heat stress are major challenges to sustainable agriculture and global food security. Here we present a hierarchically structured living material composed of directionally guided mycelium fibres, designed to regulate soil moisture and thermal load by harvesting atmospheric water and dissipating heat. The material is fabricated by cultivating Pleurotus ostreatus on a cellulose scaffold under asymmetric gas exposure, which directs hyphal growth into a vertically organized architecture with a fused cellulose–mycelium composite below and a porous aerial mycelium network above. This configuration creates a built-in wettability gradient. Aerial mycelium fibres become hydrophobic through the self-assembly of hydrophobins during upward growth into the air, whereas cellulose fibres and submerged mycelium remain hydrophilic. This gradient enables directional water transport, driving condensed atmospheric moisture from the aerial surface into the soil. Simultaneously, the porous aerial network back-scatters solar radiation and emits thermal radiation, lowering the surface temperature and promoting water vapour condensation. Field trials show that this biologically grown soil envelope increases tomato yield by approximately 28% (wet weight) compared to bare soil. Overall, this work demonstrates how the guided biomanufacturing of living materials can be harnessed to create functional, scalable and sustainable solutions for climate-resilient agriculture. This study introduces a scalable, hierarchically structured living mycelium–cellulose material that passively harvests atmospheric water and dissipates heat to regulate soil moisture and temperature, as demonstrated by increased crop yields in field trials.
|
|
Scooped by
mhryu@live.com
June 16, 11:13 PM
|
H-NS is an abundant nucleoid-associated protein found in Enterobacterales species. Some conjugative plasmids encode H-NS homologues, which are thought to facilitate plasmid maintenance and reduce the fitness costs associated with plasmid carriage. Here, we characterize HppXCROD2, an H-NS homologue encoded by the IncX4 plasmid pCROD2 of Citrobacter rodentium. Our data indicate that HppXCROD2 has a strong preference for binding pCROD2 over the chromosome or other plasmids. By contrast, chromosomally encoded H-NS displays no preference for plasmid sequence. When expressed from a heterologous plasmid in E. coli, HppXCROD2 showed similar DNA-sequence preference to chromosomally encoded H-NS. Moreover, HppXCROD2 binding to a sequence from pCROD2 was much lower when that sequence was cloned in a laboratory plasmid. Thus, HppXCROD2 preferentially binds DNA in the context of the plasmid where it is encoded, a phenomenon we term “cognate plasmid specificity”. We propose that cognate plasmid specificity occurs through recognition of plasmid-specific DNA topology generated by plasmid-encoded topoisomerases. Cognate plasmid specificity may insulate regulation of plasmid genes from the effects of host DNA, while minimizing disruption of host chromosome regulation due to plasmid carriage.
|
|
Scooped by
mhryu@live.com
June 16, 10:45 PM
|
Spores are the primary means of fungal reproduction, contributing to genetic diversity, colonization, and adaptation. Although spore formation is a pivotal part of the fungal life cycle, its genetic underpinnings remain poorly known. In this study, we characterize transcriptomic changes from late meiosis to early basidiospore formation in the mushroom-forming fungus Coprinopsis cinerea, decipher several cellular processes, and identify novel genes involved in this process. We identify distinct trajectories of gene expression, each of which display different functional signals, corresponding to meiotic and morphogenetic processes and transitions between these. Our analyses identify diverse arrays of fungal cell wall modifying carbohydrate-active enzymes, ferritins, a putative catechol-melanin synthesis pathway, as well as components of the mitotic/meiotic apparatus. We present twelve highly conserved genes with roles specific to sexual sporulation in both budding yeast and C. cinerea, indicating deep conservation of the gene networks driving sexual spore formation. Reverse genetics identified three conserved but functionally poorly characterized genes conferring sporeless and spore-poor phenotypes that result from postmeiotic developmental arrests stemming from spore inflation and nuclear migration problems. Overall, this study provides novel insight into basidiomycete spore formation and highlights the cornucopia of novel functions encoded by functionally dark genes.
|
|
Scooped by
mhryu@live.com
June 16, 9:59 PM
|
With the growing use of machine-learning-assisted pipelines for designing, characterizing, and optimizing biomolecules, the reliability of structure prediction models is increasingly important. PolyFold is a benchmarking framework developed to evaluate open-use structure prediction models, Boltz-2 and OpenFold 3, as commercially accessible alternatives to AlphaFold 3. We outline an end-to-end workflow automation tool to streamline input file creation, batch automation, and comprehensive analysis of model outputs for leading open-use structure prediction models. We curated an evaluation dataset of several thousand high-quality Protein Data Bank structures, homology-filtering against the training sets of both models to ensure a fair analysis. We then implemented an evaluation pipeline incorporating structural metrics (RMSD, TM-score, lDDT, etc.), interface metrics (DockQ, ilDDT, iRMSD, etc.), and physicochemical realism checks (based on bond lengths, angles, molecular internal energies, etc.). We identify key performance disparities, observing that Boltz-2 is generally superior to OpenFold 3, though the differential is partially attributable to residual homology leakage not accounted for by prevailing test set curation practices. We thus recommend a new method for homology-reducing when building a test set using length-weighted average fractional identity cutoffs rather than lowest chain fractional identity cutoffs. Even in eliminating residual leakage, Boltz-2 still performs better on full-set comparisons and a variety of important partitions (nucleic acids, protein-ligands, Ab-Ags, etc.). Both models are strong at folding monomeric structures, though struggle with homomultimer placement and small molecule physical realism, demonstrating enduring limitations of machine learning methods. This work is the first end-to-end, open-use, and reproducible platform for systematically assessing state-of-the-art structure prediction models. PolyFold enables practitioners to determine how models compare in performance on specific inference tasks and supports the broader adoption of accessible computational tools to facilitate biomolecular science.
|
|
Scooped by
mhryu@live.com
June 16, 9:48 PM
|
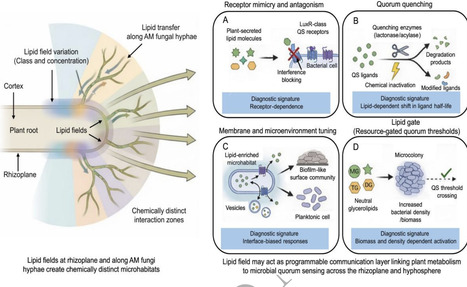
Root exudates comprise a diverse mixture of non-polar and amphiphilic compounds that are only partially recovered by aqueous extraction methods, yet can rival polar metabolites as carbon sources for microorganisms. Because many quorum-sensing (QS) signals are fatty-acyl derivatives, lipid-rich microhabitats at the root–soil interface are likely to influence signal partitioning, persistence, and local QS thresholds. We propose a lipid-mediated framework in which plant-derived lipids modulate QS through four nodes: receptor mimicry/antagonism, quorum quenching, membrane/microenvironment regulation, and lipid-dependent resource gating. These mechanisms operate across two lipid-rich interfaces - the rhizosphere and the arbuscular mycorrhizal fungi (AM fungi) hyphosphere - where host lipid fluxes may restructure microbial community composition. Combined with QS-sensitive changes in root exudation, this spatially structured lipid circuit could generate feedbacks influencing microbiome composition and function, with potential implications for microbiome engineering.
|
|
Scooped by
mhryu@live.com
June 16, 1:02 AM
|
Recombination can generate new or improved proteins by merging pieces of pre-existing genes into a new whole. Here, when we reanalyzed genomic data from several laboratory evolution experiments with E. coli, evidence for the convergent evolution of chimeric genes became apparent. In these experiments, a pair of paralogous genes were recombined into a single hybrid copy by large genomic deletions. In one case, the excision of a cryptic e14 prophage occurred in 6 out of 8 replicates of a 22-day fluoroquinolone-resistance evolution experiment. These parallel prophage excisions recombined an icd isocitrate dehydrogenase gene with a homologous icdC C-terminal fragment pseudogene. In the other case, convergent 23 kB deletions recombined cpsG paralogs across replicate populations of the Lenski long-term evolution experiment with E. coli (LTEE), generating a chimeric phosphomannomutase. Together, these results indicate that chimeric genes evolve rapidly in bacteria and are more common than previously believed, because they are easily missed by standard genomic analyses. Experiments are needed to determine whether these chimeric proteins are adaptive, are by-products of adaptive genomic deletions, or both.
|
|
Scooped by
mhryu@live.com
June 16, 12:37 AM
|
Spatial genome organization plays a crucial regulatory role, but its evolutionary development remains unclear. Leveraging Hi-C data from 1,025 species, we trace the evolutionary trajectories of genome organization through 2 higher-order architectures, “global folding” (spatial organization of the karyotype) and “checkerboard” (spatial organization of chromatin compartments). Earlier unicellular life forms mostly displayed random genome configurations. Throughout the evolution of plants, global folding became and remained the prominent architecture. However, animals progressively developed more pronounced checkerboard architectures; these are also apparent during early embryogenesis, which suggests that they act as a conserved mechanism of gene regulation. In contrast, plants exhibit comparatively weaker checkerboard patterns and instead preferentially organize co-regulated genes into linear genomic clusters. Both strategies of gene arrangement reinforce the biological principle that “structure determines function”: divergent evolutionary paths converge on architectural solutions that reflect gene regulatory requirements over time.
|
|
Scooped by
mhryu@live.com
June 16, 12:27 AM
|
Emerging fungal pathogens represent a concerning threat to both global health and food security. In this study, we aimed to address our rising vulnerability to fungal pathogens through the development of the Fung-AI pipeline: an AI/ML-driven approach for antifungal discovery. A generative adversarial network (GAN) was trained to generate novel candidate antifungal peptide sequences. Next, in silico antifungal and hemolytic classifiers were built to further prioritize AI-generated peptides for experimental validation. From a pool of ~10,000 candidates, thirteen peptides were selected for testing over two-stages of experimentation. Five peptides were found to display mild antifungal activity against the wheat pathogen, Fusarium graminearum, with minimal inhibitory concentrations (MICs) ranging from 250 µg/mL to 500 µg/mL. Four of the five peptides also showed activity against the human pathogen, Candida albicans (MIC: 500 µg/mL). Two of our AI-generated antifungal peptides additionally demonstrated low cytotoxicity in HepG2 human liver carcinoma cells (LC50 > 704.2 µg/mL) indicating that they may be useful as scaffolds for future optimization for therapeutic applications. None of our peptides were found to considerably inhibit the emerging pathogen C. auris, suggesting the need for pathogen-specific down-selection of candidate peptides. Overall, we present a proof-of-principle, generative-AI-based approach for the rapid design of de novo antifungal peptides.
|
|
Scooped by
mhryu@live.com
June 16, 12:15 AM
|
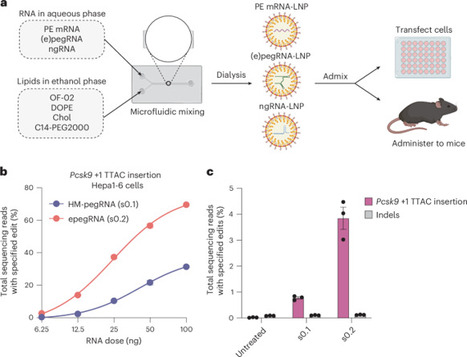
Prime editing is a versatile clinical genome editing method that enables precise substitutions, small insertions and deletions at specified locations in the genomes of living systems including human cells. Although non-viral lipid nanoparticle (LNP) delivery of RNA in vivo has become a preferred method for gene editing in animals and patients, its application to complex, three-component prime editing systems has yielded low editing efficiencies. Here we developed a systematic prime editing LNP (PE-LNP) optimization platform that addresses key bottlenecks in cargo design that limit editing efficiency. This generalizable workflow yielded PE-LNPs that can achieve 49% average in vivo prime editing in the bulk mouse liver with a single dose of 2 mg kg−1. We applied our workflow to the correction of PAH R408W, a cause of phenylketonuria, in a mouse model and achieved prime editing efficiencies and serum phenylalanine levels anticipated to be curative. We also show that PE-LNPs minimize off-target editing compared with DNA delivery methods, induce only transient elevation of liver enzymes and can be dosed repeatedly to improve editing efficiencies. These PE-LNP systems provide an attractive alternative to viral delivery by offering transient expression that minimizes off-target editing, no observed long-term toxicity and high levels of non-viral in vivo liver prime editing. A systematic workflow is used to optimize lipid nanoparticle-based prime editing systems that enable efficient editing and disease correction in human cells and mouse tissue, and overcome key limitations of transient delivery.
|







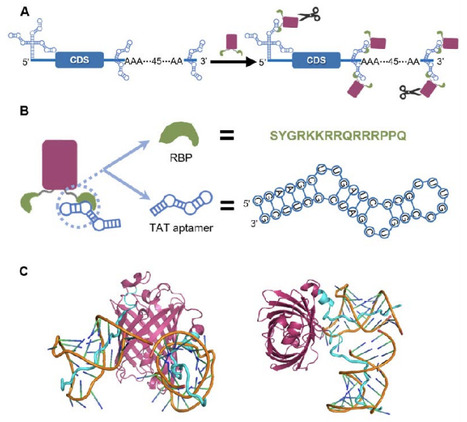
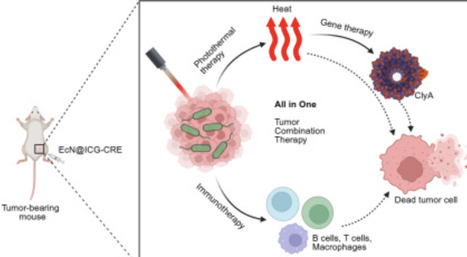


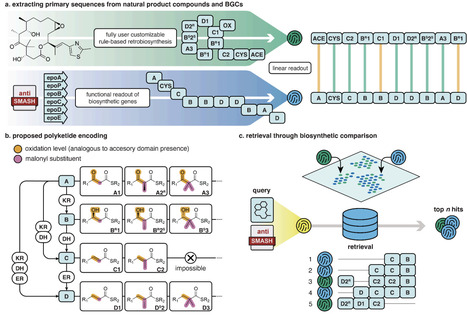
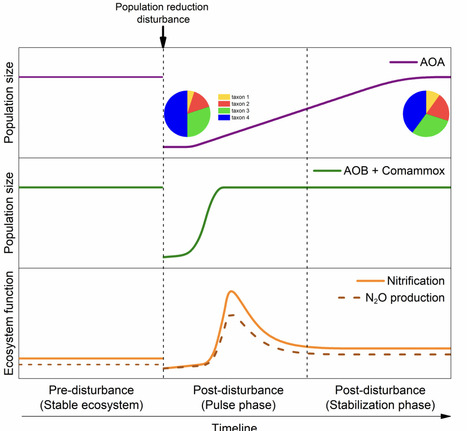










mutator ΔmutH and ΔmutT: carry different DNA repair defects that produce distinct mutational spectra.
The old assumption in large-population evolutionary biology is that when population size is large enough, selection coefficients dominate and mutational bias becomes irrelevant because any beneficial mutation will eventually arise regardless of its mutational likelihood, and the fitter variant wins.