Your new post is loading...

|
Scooped by
mhryu@live.com
Today, 12:14 AM
|
To advance the computational simulation of cellular life, we propose a virtual yeast, an artificial intelligence (AI)-driven agent that models eukaryotic cellular behaviors by integrating multimodal biological data, mechanistic reasoning and active experimentation using Saccharomyces cerevisiae as a genetically tractable and data-rich model system. Cellular complexity is decomposed into eight function-centred modules, spanning genetic, metabolic and structural systems, each realized as a domain-specific AI tool coordinated through a large language model-based orchestration layer. Built on three data pillars, namely, mechanistic knowledge, subcellular architecture and dynamic states, the system integrates representation learning and generative modelling within a closed-loop learning pipeline that autonomously designs and executes experiments. The virtual yeast serves as both a conceptual and an operational platform to optimize biosynthetic pathways, support the generation and prioritization of hypotheses across diverse cellular processes, and accelerate target discovery. By coupling biological realism with autonomous AI reasoning, the virtual yeast establishes a generalizable blueprint for constructing virtual eukaryotic cells and advancing synthetic biology. A virtual yeast, an artificial intelligence-driven agent for modelling eukaryotic cellular behaviours, is described.
|
|
Scooped by
mhryu@live.com
July 1, 11:56 PM
|
The recent revolution in genome sequencing and protein structure prediction has opened new frontiers in understanding, predicting and designing enzyme function. Central to these efforts is the discovery and functional annotation of novel enzymes, which is essential for elucidating the connection between genotype and phenotype and for developing biocatalysts for industrial applications. However, accurately predicting enzymatic function remains a major challenge, and the discovery of new enzymes often relies on serendipity. Here we present a metal-coordination-guided strategy that uses atomic-level mechanistic principles to mine protein structure databases for the targeted discovery of metalloenzymes. We apply this framework to the AlphaFold2 Protein Structure Database to identify new members of the FeII/α-ketoglutarate-dependent halogenase family, which selectively functionalize unactivated C(sp3)-H-bonds, a crucial transformation in the production of pharmaceuticals and other high-value compounds. These radical halogenases constitute a low-abundance class within the large and diverse cupin superfamily. Owing to low sequence conservation, they have been especially challenging to find against the complex background of related family members, such as hydroxylases, desaturases and epimerases. Our metal-coordination mining methodology reveals several previously unrecognized radical halogenase families spanning diverse phylogenetic space, at minimal computational cost. Our predictions are validated by the experimental characterization of two new radical halogenases, AspX and BtnX. Notably, BtnX shows a substrate promiscuity that is unprecedented in radical halogenases, opening the way for a broad range of biocatalytic applications. A methodology for mining protein structure databases on the basis of the distinct intrinsic structures of metal-binding active sites in enzymes enables the discovery of new families of radical halogenases.
|
|
Scooped by
mhryu@live.com
July 1, 11:38 PM
|
Messenger RNA levels, crucial for cell survival and adaptation, are regulated through their degradation by ribonucleases (RNases). Although the molecular mechanisms of RNases in E. coli are established, the broader effects of RNases on growth and metabolism remain unclear. Here, the roles of three RNases, E, II, and R, were examined individually and in combination in double and triple mutants. Growth behaviors and metabolic changes were analyzed on different carbon sources and under recombinant protein production conditions. C-terminal truncation of RNase E was unexpectedly found to have strong effects, promoting carbon-storage metabolism, thereby leading to glycogen accumulation, especially with glucose as a carbon source. Even more surprisingly, it accelerated growth on xylose. Synergistic interactions between the three RNases were also identified. Deleting RNase R amplified glycogen accumulation in the RNase E mutant, and further increased its growth rate. All three RNases were found to significantly contribute to acetate overflow regulation, with synergy between RNase E and R. Combined mutations had additional benefits under protein production conditions. Compared with the parental strain, the double mutant with both RNase E truncation and RNase II deletion produced up to twice as much recombinant protein, grew faster on xylose, and produced more glycogen on glucose. Overall, this work shows that RNase E, II, and R act both independently and synergistically in controlling E. coli growth and metabolism across carbon sources and bioproduction conditions. These findings highlight the strong relationship between RNA degradation and cell physiology and offer perspectives for engineering optimized microbial chassis in biotechnology.
|
|
Scooped by
mhryu@live.com
July 1, 10:01 PM
|
Synthetic mRNA therapeutics offer a versatile platform for treating diverse conditions, including cancer and infectious diseases. For delivery into cells, these mRNAs are encapsulated in lipid nanoparticles and commonly incorporate modified ribonucleotides to improve stability, enhance translation and mitigate immune recognition. N1-Methylpseudouridine (m1Ψ) has become the industry standard for synthetic mRNAs owing to its effectiveness in promoting translation and reducing immunogenicity. However, recent studies have shown that m1Ψ can compromise translational fidelity, leading to errors such as premature termination and ribosomal frameshifting. Here we reveal N4-acetylcytidine (ac4C) as a functionally distinct alternative to m1Ψ. Across cultured cell lines, primary human monocyte-derived dendritic cells and mouse liver, ac4C suppressed inflammatory responses as effectively as m1Ψ while driving higher protein yields. Single-molecule imaging of translation revealed broadly similar ribosome densities per mRNA for ac4C-modified and m1Ψ-modified transcripts. However, translation elongation with m1Ψ-modified mRNA was nearly twofold slower than with ac4C, which resulted in reduced protein output and increased ribosome collisions that further limited protein production through the engagement of quality-control pathways and +1 frameshifting. These findings underscore the importance of context in designing therapeutic mRNAs and position the translation elongation rate as a key determinant of the efficacy of modified ribonucleotides. Different RNA modifications elicit different translation elongation rates for in vitro transcribed mRNAs that result in disparate translation outputs and fidelity, as shown here for N4-acetylcytidine versus the industry standard for synthetic mRNAs, N1-methylpseudouridine.
|
|
Scooped by
mhryu@live.com
July 1, 4:58 PM
|
Metabolic engineering efforts are determined not only by enzyme activity but also by the thermodynamic driving force. This forum argues that many ‘novel’ synthetic routes merely relocate energetic penalties rather than eliminate them and demonstrates why evaluating Gibbs free energy change under physiological conditions is essential for robust, optimal pathway design.
|
|
Scooped by
mhryu@live.com
July 1, 4:40 PM
|
Since its identification two decades ago, the T6SS has emerged as far more than a pedestrian homologue of the T4SS. What began as a curiosity in pathogenesis research has revealed itself to be a central player in microbial ecology, mediating interactions that range from symbiotic colonization to outright predation. The system’s remarkable diversity in structure, regulation and deployment reflects the breadth of ecological contexts in which bacteria have co-opted this molecular weapon. As researchers continue to probe the evolutionary and ecological consequences of T6SS activity in complex communities, this system promises to illuminate fundamental principles of bacterial warfare and cooperation that shape the microbial world.
|
|
Scooped by
mhryu@live.com
July 1, 4:35 PM
|
Pantothenic acid (PA), or vitamin B5, can be synthesized by gut commensals, but the contribution of microbial PA to metabolic health remains unclear. Here, we find that microbial PA supply is reduced in individuals with metabolic syndrome (MetS) and is associated with impaired gut barrier function and disease severity. Tracing microbial PA identifies Bacteroides fragilis as a key contributor, with panC required for PA biosynthesis, as confirmed by isotope tracing, bacterial culture, and germ-free colonization. In MetS models, colonization with wild-type, but not ΔpanC B. fragilis, restores PA, preserves gut barrier integrity, reduces endotoxemia, and improves metabolic dysfunction. Mechanistically, microbial PA requires host pantothenate kinase activity, as silencing pantothenate kinase 2/3 (PANK2/3) in colonic organoids and in vivo reduces coenzyme A (CoA)/acetyl-CoA metabolism, suppresses Krüppel-like factor 4 (KLF4)-associated differentiation programs, and blunts the protective effects of microbial PA. Finally, a plant-derived polysaccharide enriches PA-producing Bacteroides and restores colonic PA, highlighting a strategy for colonic homeostasis and metabolic health.
|
|
Scooped by
mhryu@live.com
July 1, 3:18 PM
|
Protein sequence alignment is a crucial task in bioinformatics, yet aligning remote homologs with low sequence identity remains a longstanding challenge, particularly due to the difficulty of handling gaps. We introduce a new method that applies Optimal Transport (OT) theory to sequence alignment, providing a mathematically principled framework for modeling residue matches and gaps. OTalign formulates sequence alignment as an entropy-regularized unbalanced optimal transport (UOT) problem over embeddings derived from protein language models (PLMs). Unlike traditional methods, it introduces position-specific gap penalties that adapt to each sequence pair. On challenging remote-homolog benchmarks (SABmark, MALIDUP, MALISAM), OTalign consistently outperforms baselines (Needleman-Wunsch, HHalign) and recent PLM-based methods (PLMAlign, DeepBLAST), achieving F1 scores of 0.594 on SABmark Superfamily and 0.358 on SABmark Twilight. Furthermore, OTalign provides a quantitative and interpretable metric of how effectively PLM embeddings represent sequence similarity relationships. Finally, its differentiable nature enables end-to-end fine-tuning of PLMs, establishing a framework for learning embeddings explicitly optimized for alignment tasks.
|
|
Scooped by
mhryu@live.com
June 30, 11:48 PM
|
Accurate species-level classification of prokaryotic 16S rRNA sequences remains difficult: existing tools rely on exact alignment, k-mer heuristics, or phylogenetic placement and are limited by incomplete reference databases. Deep learning approaches in microbial genomics have focused largely on whole-genome metagenomics, leaving 16S taxonomy under-supported. We present DeepTaxa, a hybrid CNN-BERT framework that pairs a multiscale CNN with a transformer trained from scratch on the DNABERT-2 BPE vocabulary, producing parallel rank-specific predictions across the seven Linnean ranks. On the Greengenes2 2024.09 test set, DeepTaxa achieves species-level accuracy of 92.96% and F1 of 0.9212 (3-seed mean; cross-seed standard deviation F1 at every rank), with F1 above 0.99 from domain through class and a species-level expected calibration error of 0.0242. DeepTaxa exceeds DADA2 (90.05%) and QIIME 2 (85.01%) at the species rank on the same held-out test set, with larger gains over the k-mer-based classifiers SINTAX and Kraken 2. Performance degrades smoothly with decreasing training-set similarity (species F1 from 0.95 to 0.45), and a dedicated V3–V4 amplicon checkpoint reaches 87.55% species accuracy from an approximately 420 bp window.
|
|
Scooped by
mhryu@live.com
June 30, 10:49 PM
|
Biosynthetic gene clusters (BGCs) are contiguous genomic regions that encode diverse proteins responsible for natural product biosynthesis. These proteins collectively produce various secondary metabolites with complex chemical structure, including antibiotics and mycotoxins, yet the complete biosynthetic pathways have been experimentally elucidated for only a limited number of compounds. Recently, protein–protein interactions within BGCs have been recognized as key determinants of intermediate transfer, enzymatic regulation, and structural stability. However, many BGCs still contain proteins of unknown function that cannot be predicted by conventional sequence-based bioinformatics tools, hindering a comprehensive understanding of their biosynthetic pathways. To address this challenge, we built a high-throughput complex prediction pipeline by replacing AlphaFold3’s multiple sequence alignment generation with a faster MMSeqs2. We systematically screened 487,828 protein pairs derived from 2,437 BGCs registered in the Minimum Information about a Biosynthetic Gene cluster database and predicted 15,438 heteromeric interactions with an interface predicted TM score ≥ 0.6. Among them, 1,390 protein pairs exhibited structural homology with a root mean square deviation ≤ 2.0 Å. Our analysis demonstrates that AF3-based complex prediction matches experimental results with high confidence for most proteins and reveals many uncharacterized but novel heterocomplexes within BGCs. These findings will facilitate experimental verification of unidentified enzymatic reactions leading to the final products.
|
|
Scooped by
mhryu@live.com
June 30, 3:13 PM
|
Prime editing has not been established in filamentous fungi, which are major ecological contributors and industrial hosts with vast biosynthetic capacity. Here we develop fPE7max, a prime editing platform optimized for fungi, which supports different edit types, including base substitutions and defined small insertions or deletions, with an average editing efficiency approaching 90%, across diverse genomic loci and species. fPE7max further enables larger insertions of up to 1 kb and deletions of up to 10 kb. We perturb upstream open reading frames in the pleiotropic regulator gene, laeA, to modulate metabolic output across multiple fungal species. Metabolomic profiling reveals activation of previously lowly biosynthetic pathways, leading to the identification of 18 metabolites, including 8, to our knowledge, previously unreported structures, 3 of which with cytotoxic activity. These results establish fPE7max as an efficient platform for genome engineering in filamentous fungi and show upstream open reading frame editing as a strategy for modulating endogenous regulatory networks and accessing the fungal chemical repertoire. fPE7max efficiently installs base substitutions, insertions and deletions to modulate filamentous fungal metabolism.
|
|
Scooped by
mhryu@live.com
June 30, 2:43 PM
|
Protein-protein interactions (PPI) are central to biological processes. Designing small molecules that modulate dysregulated PPIs holds strong promise for targeting undruggable proteins. However, existing structure-based drug design approaches focus on well-defined small-molecule binding pockets and struggle to generalize to large, shallow, and chemically complex PPI interfaces. Here, we introduce Pep2Mol, a diffusion-based generative model for 3D molecule design that targets orthosteric PPI sites by explicitly incorporating binding peptides or proteins as structural guidance, moving beyond conventional pocket-conditioned generation. To enable model development and benchmarking, we curate a large-scale, high-quality dataset of 10,956 experimentally resolved protein complex structure pairs, each pairing an orthosteric competitive ligand with a protein binder at overlapping receptor interfaces. Pep2Mol integrates two SE(3)-equivariant graph neural networks that encode protein-ligand and protein-peptide interactions respectively, and fuses these representations via attention-based conditioning to jointly guide the diffusion trajectory. Extensive evaluations demonstrate that Pep2Mol generates chemically valid ligands with state-of-the-art binding affinities, providing a strong foundation for small-molecule inhibitor design against challenging PPI interfaces.
|
|
Scooped by
mhryu@live.com
June 30, 1:58 PM
|
Metalloproteins are essential to many cellular processes. They use metal ions as cofactors to catalyze reactions, stabilize protein structures, and mediate electron transfer. Identifying their metal-binding sites remains difficult because of the complexity of protein environments and the promiscuous binding of metal ions, and existing computational methods are limited by accuracy and data scarcity. Here we introduce PRIME, a hybrid deep learning framework that combines evolutionary and structural signals to predict metal-binding sites accurately and efficiently. PRIME employs protein language models and pre-trained structure models to extract information from protein sequences and structures, together with a probe generation algorithm that bridges sequence- and structure-based predictions by scanning candidate sites. PRIME outperforms existing methods across diverse metal ions, from abundant zinc and calcium to challenging potassium and sodium. Ablation analysis shows that pretrained structure models improve accuracy. Case studies on AlphaFold2 models further demonstrate PRIME’s potential for high-throughput metalloproteomics. The Authors present PRIME, a deep learning tool that fuses protein sequence and structure to pinpoint metal-binding sites across diverse metal ions, from abundant zinc to rare potassium, offering improvements in speed and accuracy over existing methods.
|
|
|
Scooped by
mhryu@live.com
Today, 12:05 AM
|
Intracellular bacterial symbioses have arisen myriad times in eukaryotes, with dozens known from insects alone. Beginning with Buchnera, the obligate endosymbiont of aphids, genomes of endosymbionts have illuminated their evolutionary origins and metabolic contributions to hosts. However, the mechanisms by which non-culturable endosymbionts enter host cells and suppress cellular immune processes have remained unclear. Here we show that an uncharacterized Buchnera protein, designated SyeA, was present in the Buchnera ancestor, is secreted into the host cytoplasm, is homologous to secreted effectors of bacterial pathogens and is essential for Buchnera transmission. Buchnera is transmitted through expulsion from specialized maternal cells and uptake by embryos. Using immunofluorescence microscopy, we found elevated SyeA levels after colonization of the embryonic cell, accompanied by actin accumulation at the entry site. SyeA localizes outside the host-derived membrane and actin layer surrounding each Buchnera cell within host cytoplasm. Knockdown of syeA expression disrupts colonization of embryos and embryonic development and elevates lysosomal activity, leading to Buchnera destruction. Our findings provide insights into how an anciently associated, mutualistic endosymbiont achieves its intracellular existence. SyeA represents a vestige of pathogenic origins that was followed by evolution of increased host control and erosion of the original, more complex pathogenicity machinery. SyeA was present in the Buchnera ancestor, is secreted into the host cytoplasm, is homologous to secreted effectors of bacterial pathogens and is essential for Buchnera transmission.
|
|
Scooped by
mhryu@live.com
July 1, 11:39 PM
|
A Chemically Defined Synthetic Cell Capable of Growth and Replication. Natural cells divide using internal scaffolding called a cytoskeleton. Building a functional cytoskeleton from scratch has been a major bottleneck in synthetic cell research because it requires dozens of proteins working in coordination. SpudCell sidesteps this entirely, with proteins crowding together on the membrane surface until the mechanical stress makes the membrane split. Cells that make more of this surface protein divide more efficiently, directly coupling the genome to reproductive success.
|
|
Scooped by
mhryu@live.com
July 1, 11:17 PM
|
Aminobacter niigataensis MSH1 is a candidate for bioaugmentation of sand filters in drinking water treatment plants (DWTP), as it mineralizes the ubiquitous groundwater micropollutant 2,6-dichlorobenzamide (BAM). The DWTP sand filter isolate Piscinibacter sp. K169 improves BAM mineralization by MSH1 in an apparent accidental mutual cooperation, and co-inoculation of the organism was proposed to assist bioaugmentation with MSH1. In this study, we questioned whether this accidental mutual positive interaction extends to four other pesticide catabolic bacterial strains of the same or a different genus of MSH1, and examined the longevity of the cooperation. Negative interactions were never observed in either direction. As observed for BAM mineralization by MSH1, K169 stimulated BAM mineralization by A. niigataensis LG1 and 2,4-D mineralization by Cupriavidus pinatubonensis JMP134 without affecting the cell density of the catabolic strains. Linuron mineralization by Variovorax sp. SRS16 and carbofuran mineralization by Novosphingobium sp. KN65.2 were not affected. In the other direction, growth of K169 was stimulated by all pesticide catabolic strains except JMP134, indicating a common underlying mechanism. After 2 weeks, the beneficial effects of K169 on MSH1, LG1, and JMP134 functionality diminished or even reversed, likely because of organic carbon depletion. In contrast, cell densities of K169 in all dual-species systems remained higher than in the K169 monoculture system. This study extends our knowledge on accidental interactions and the beneficial effect of a sand filter isolate toward other pesticide degraders, opening doors for Piscinibacter sp. K169-assisted bioaugmentation of other/multiple pesticide degraders in DWTPs.
|
|
Scooped by
mhryu@live.com
July 1, 5:01 PM
|
The KARRIKIN INSENSITIVE 2 (KAI2) receptor has been reported to contribute to drought tolerance in Arabidopsis. However, the extent to which KAI2’s function in drought tolerance depends on soil microbiota remains unclear. This study demonstrates that the rhizosphere microbiome is indispensable for KAI2-mediated drought tolerance. We isolated specific Rhodanobacter sp. and confirmed its role in enhancing drought tolerance in Arabidopsis. Notably, Rhodanobacter sp. was found to specifically secrete the key isoflavone daidzin. We found that daidzin had a similar function with KAI2 agonist, desmethyl-type germinone, and induced interaction between KAI2 and SUPRESSOR OF MORE AXILLARY GROWTH 2 1. Moreover, the exogenous application of daidzin enhanced drought tolerance by modulating the expression of karrikin response and drought-related genes, in a KAI2-dependent manner. Our findings suggest that the rhizosphere microbiome plays a crucial role in facilitating KAI2-mediated drought tolerance in Arabidopsis, with Rhodanobacter sp. contributing through the secretion of daidzin.
|
|
Scooped by
mhryu@live.com
July 1, 4:55 PM
|
C1 compounds are abundant, non-food and renewable feedstocks, making them attractive substrates for producing value-added chemicals via microbial bioconversion. In nature, autotrophic microorganisms assimilate C1 substrates, including CO, CO2, methane, methanol and formate, through native C1 fixation and assimilation pathways. Building on these natural routes, synthetic C1 assimilation pathways and engineered microbial cell factories have improved C1 utilization and broaden product portfolios. This review presents the recent progress and current strategies in producing high-value compounds using microbes possessing natural and non-natural C1 assimilation modules. We highlight key bottlenecks that limit efficient C1 assimilation and discuss potential strategies to address them, outlining opportunities for future C1-based biomanufacturing.
|
|
Scooped by
mhryu@live.com
July 1, 4:39 PM
|
Chemical modification of oligonucleotides has become an essential strategy to improve their properties and expand their functional utility in chemical biology, molecular imaging, and therapeutics. Among the various modification strategies, post-synthetic bioorthogonal click chemistry enables efficient, rapid, and biocompatible site-specific conjugation of functional moieties. Central to these advances is the development of clickable nucleoside phosphoramidites, namely nucleoside building blocks containing reactive bioorthogonal click handles that are compatible with the harsh conditions of solid-phase oligonucleotide synthesis (SPOS). This review provides a comprehensive overview of bioorthogonal cycloaddition-based nucleoside phosphoramidites reported to date that are compatible with SPOS. We highlight their synthetic accessibility and deviations from conventional SPOS protocols when required, as well as the stability and hybridization behavior of the resulting clickable oligonucleotides. Furthermore, we examine the performance of subsequent bioorthogonal reactions used for post-synthetic functionalization, with particular attention to their kinetics and efficiencies. Representative applications are also discussed, including the development of labeled probes, multifunctional assemblies, and targeted delivery systems.
|
|
Scooped by
mhryu@live.com
July 1, 3:51 PM
|
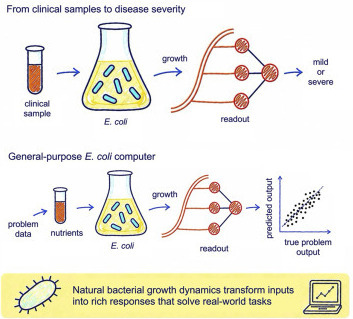
We introduce a systems-level approach to sensing and computing in which E. coli acts as a living reservoir computer, performing complex information processing through its native growth responses without requiring genetic modification or specialized instrumentation. We validate this framework by accurately classifying early-stage COVID-19 plasma samples according to subsequent disease severity using only bacterial growth data, highlighting its prognostic potential without the need for infrastructure-dependent methods. By controlling nutrient media compositions, we also demonstrate that E. coli growth encodes nonlinear transformations that outperform linear regression, support vector machines, and multilayer perceptrons across diverse regression and classification tasks. More broadly, simulations across genome-scale metabolic models from multiple bacterial species support a link between phenotypic diversity and computational capacity. These findings position biological reservoir computing as a robust, scalable, and low-cost platform for intelligent biosensing, diagnostics, and hybrid bio-digital computation, while providing new mechanistic insights into the computational capabilities of living systems.
|
|
Scooped by
mhryu@live.com
July 1, 12:08 AM
|
The limestone monuments of the Rectangular Tower in the Xiaoling Tomb of the Ming Dynasty, created in the mid-fourteenth century, are biodeteriorating from environmental exposure, resulting in the formation of black biocrusts. However, the microbiomes that shape biocrust formation and the biodeterioration processes involved remain unclear, significantly challenging the conservation of stone monuments at this archaeological site. Here, we systematically investigated the physicochemical properties and microbial communities of biocrusts to identify keystone taxa that shape their formation and biodeterioration. Physicochemical analysis indicated that biological crusts are associated with calcium mobilization and redistribution of the limestone monuments. Microscopy and spectroscopy indicated that microbial interactions with limestone promote the formation of biological crusts. Importantly, we observed the significant predominance of Cyanobacteria and/or Chloroflexi in biocrusts, suggesting that photosynthesis may be a crucial process in biocrust formation. Fungal communities in biocrusts were dominated by Ascomycota, Basidiomycota, and Chytridiomycota, while archaeal communities were dominated solely by Nitrososphaerota. Microbial co-occurrence network and correlation analyses identified 12 keystone taxa across 11 genera that shape biocrust formation. Importantly, Scytonema spp. could provide organic carbon and nitrogen for Spirosomaceae spp., and members of the classes Cyanobacteriia and Agaricomycetes, as well as the genera Setophaeosphaeria and Plectosphaerella, are likely the keystone taxa responsible for both biocrust formation and the associated biodeterioration. Additionally, two predominant ammonia-oxidizing archaeal families (i.e., Nitrososphaeraceae and Candidatus Nitrocosmicus) could support chemolithoautotrophic growth in the microbiome by oxidizing ammonia and fixing carbon dioxide. Together, these findings underscore the need for targeted conservation strategies to mitigate microbial biodeterioration of stone monuments during biocrust formation.
|
|
Scooped by
mhryu@live.com
June 30, 11:08 PM
|
Understanding pathogen metabolism is critical for identifying key functions for drug targeting, establishing effective in vitro experimental systems, particularly for metabolically unique organisms such as Leptospira. Pathogenic Leptospira are thought to infect humans from environmental sources; however, direct isolation from environmental samples remains technically challenging and is not yet well established. Here, we report that a ubiquitous environmental bacterium belonging to the former Massilia group produces metabolites to promote the growth of Leptospira interrogans, which has been encountered through an incidental contamination event and analyzed in this study. Gas chromatography–tandem mass spectrometry (GC-MS/MS) analysis demonstrated that cultivation of Massilia sp. strain NBRC 108631 in R2A medium resulted in the accumulation of metabolites, including branched-chain amino acid (BCAA) intermediates, compared to fresh medium. By combining genome-scale metabolic modeling with experimental validation using cell-free culture supernatant supplementation assays, we demonstrate that BCAA intermediates, particularly 2-ketoisocaproic acid (4-methyl-2-oxopentanoate; 4MOP), a leucine biosynthetic intermediate produced by strain NBRC 108631, enhance Leptospira growth. To investigate the metabolic role of 4MOP, we incorporated transcriptomic data into a genome-scale metabolic network model to generate condition-specific models. Resulted flux distributions indicated that Leptospira catabolized imported 4MOP to produce acetyl-CoA. Our results reveal a previously unrecognized metabolic interaction where metabolites produced by environmental bacteria support the growth of pathogenic Leptospira, offering mechanistic insight into its metabolic requirement. These findings have implications to understand the environmental persistence of Leptospira through its metabolic dependencies on coexisting microbes, and they also help develop better strategies for this pathogen.
|
|
Scooped by
mhryu@live.com
June 30, 3:35 PM
|
Pathogens deploy effector proteins to exploit host cell biology, and most effector open reading frames (ORFs) are rapidly evolving and lack functional annotation. We developed the effector ORFeome (eORFeome), a scalable functional genomics platform encompassing 3,835 effector ORFs from diverse viruses, bacteria, and parasites. High-throughput barcoded screens across nuclear factor κB (NF-κB), apoptosis, p53, cGAS-STING, and major histocompatibility complex class I (MHC class I) pathways revealed novel pathway-modulating functions for hundreds of uncharacterized eORFs, unexpected activities of known effectors, and distinct pathway-specific functions encoded by single ORFs. Illustrating the power of this approach, we identified HHV6A U14 as a p53 antagonist, HHV7 U21 as a dual-function STING antagonist and MHC-I antigen display inhibitor, and adenoviral 13.6K/i-leader protein as a de novo-evolved TAP inhibitor that suppresses MHC-I display. These results establish a general framework for systematic effector annotation, uncover new mechanisms of host-pathogen interaction across kingdoms, and highlight pathogen effectors as a versatile toolkit for rewiring and probing human cellular pathways.
|
|
Scooped by
mhryu@live.com
June 30, 2:49 PM
|
Germinal is a recently described computational pipeline for de novo antibody design that combines AlphaFold-Multimer hallucination with antibody language model guidance to generate epitope-targeted antibodies. Germinal identified binders with nanomolar-to-low-micromolar affinities by testing only 43-101 designs per target across four diverse antigens, establishing it as a practical tool for epitope-directed antibody design accessible to standard academic laboratories. As this architecture is itself very recent, systematic replacement and benchmarking of its individual components remains largely unexplored, yet offers a valuable opportunity to probe the robustness of the underlying design. We present OpenGerminal, which replaces PyRosetta with a fully open-source stack comprising OpenMM 8.5.1, FreeSASA, FASPR, Biopython, and sc-rs v1.0.0, and adopts AbLang1 (ablang2 v0.2.1) as the sole antibody language model in place of IgLM. Benchmarking on two VHH targets (PD-L1 and IL-3) reveals that OpenGerminal achieves a markedly higher cofolding pass rate (PD-L1: 33.7% vs. 18.6%; IL-3: 24.6% vs. 8.0%) with equivalent or improved Chai-1 structural confidence metrics in accepted designs, at the cost of a modest increase in per-trajectory computation time (>=1.5x). Multi-chain target support is also extended and verified to run without error on the official insulin example. OpenGerminal provides the first systematic benchmarking of IgLM versus AbLang1 within the Germinal architecture, and its fully open-source component stack broadens the range of deployment contexts in which the pipeline can be used.
|
|
Scooped by
mhryu@live.com
June 30, 2:32 PM
|
High-throughput screening of protein domains enables the systematic discovery of protein sequences that encode specific cellular functions. Fluorescence-activated cell sorting-based assays have long been the standard readout for such screens but remain time- and resource-intensive, imposing practical limits on library size and coverage. Here we describe a scalable magnetic separation-based workflow that provides an alternative to FACS for screening large protein libraries in mammalian cells. We engineered a modular synthetic surface marker, consisting of a fusion between the fragment crystallizable (Fc) region of human immunoglobulin G and the transmembrane domain of platelet-derived growth factor receptor-β, that allows cells to be magnetically separated on the basis of surface reporter expression using Protein G-coated magnetic beads. The procedure covers pooled library cloning, lentiviral delivery, magnetic separation and sequencing-based quantification, enabling reproducible screening of more than 100,000 protein domain variants. The approach is suitable for the identification of functional protein domains capable of transcriptional and post-transcriptional RNA regulation and may lead to the selection of improved transmembrane domains for efficient protein surface display. The entire workflow, from library design to data analysis, can be completed in 4–6 weeks and requires skills in cell culture, molecular cloning and computational techniques. This scalable and accessible Protocol enables researchers to systematically measure protein domain functions across biological contexts, thus accelerating both biological discovery and protein engineering. This Protocol presents a scalable magnetic separation-based method for screening large protein domain libraries in mammalian cells, providing a simple and quantitative alternative to fluorescence-activated cell sorting for functional protein discovery.
|